












HTML
-
The dengue virus (DENV) is a single-stranded positive-sense RNA virus that belongs to the family Flaviviridae (Gubler, 2002), and has four serotypes, DENV1-DENV4, which are transmitted via Aedes aegypti and Aedes albopictus (Rodriguez-Roche and Gould, 2013). It has been reported that more than 50 million dengue infections occur each year (Guzman et al., 2010), and a serious outbreak occurred in the Southern Provinces of China in the summer of 2014. The clinical presentations of dengue infection range from mild febrile illness to severe dengue characterized by dengue hemorrhagic fever and shock syndrome, which make the accurate laboratory confirmation of the diagnosis challenging but crucial. Currently, serological assays and real-time quantitative polymerase chain reaction (qPCR) techniques are most commonly applied in the diagnosis of dengue infection (Guzman et al., 2010; Bäck and Lundkvist, 2013; Guzman and Harris, 2015). However, the current methods have some major drawbacks, such as false-positive results owing to cross-reactivity with other ar-thropod-borne flaviviruses, and inability to determine the infectiousness in individual patients (Schwartz et al., 2000). Therefore, a simple and accurate method is needed to detect the virus, especially its associated infectious particles.
Propidium monoazide (PMA) is a fluorescent, photo-reactive dye with a high affinity for nucleic acids. Previous studies have indicated that PCR combined with PMA can discriminate between viable and inactivated cells, because of the reduction in PCR signal from DNA of dead cells (Nocker et al., 2006). The azide group of the compound generates highly reactive nitrene intermediates that readily form covalent nitrogen-carbon bonds with nucleic acids. The nucleic acids modified by PMA cannot be used as templates to be transcribed or amplified. However, the compound does not penetrate intact cell walls or membranes, and only reacts with DNA or RNA exposed after such barriers are damaged (Nocker et al., 2006). Hence, the compound may be used to selec-tively amplify templates from structurally intact or-ganisms. The PMA-qPCR method has been widely applied to detect and quantify viable particles of the echovirus (Parshionikar et al., 2010), bacteriophage T4 (Fittipaldi et al., 2010), influenza virus (Graiver et al., 2010), and norovirus (Kim and Ko, 2012). In this study, we investigated the feasibility and reliability of selective qPCR to detect DENV2.
Cytopathogenic DENV2 was obtained from the BSL-3 Laboratory at the Southern Medical University, China, and was propagated and assayed in BHK-21 cells. Semipurified stocks were subsequently produced from these cultures. Briefly, the cells were lysed by repeated freeze-thaw cycles at -80 ℃ (Low-temperature Refrigerator, SANYO, Osaka, Japan); the lysates were cleared by centrifugation for 15 min at 4000 rpm at 4 ℃, and then stored at -80 ℃. The infectious titers were measured as a 50% tissue culture infectious dose in eight replicates. Clustal W was used to align the specific conserved cDNA fragment from 20 DENV2 strains isolated from different geographical areas at different times. The primers for this fragment were designed by using the Primer 5.5, and assessed by using the Oligo 7.0 program (Supplementary Table S1). The fragment was also synthesized, inserted into the plasmid pMD18-T (TAKARA Co., Dalian, China) to construct the standard plasmid pMD-DENV2(Figure 1A). The plasmids were propa-gated in Escherichia coli DH5α and purified by using the TAKARA MiniBEST plasmid purification kit. A stan-dard curve was generated by amplifying 10-fold dilutions of the standard plasmid (Supplementary Figure S1A), starting with a stock concentration of 279.1 ng/µL, and then the cycle threshold (Ct) values were plotted against the concentrations of the template [Supplementary Figure S1B, Y = -3.258 × Log (X)± 9.79, Eff. = 102.7%].
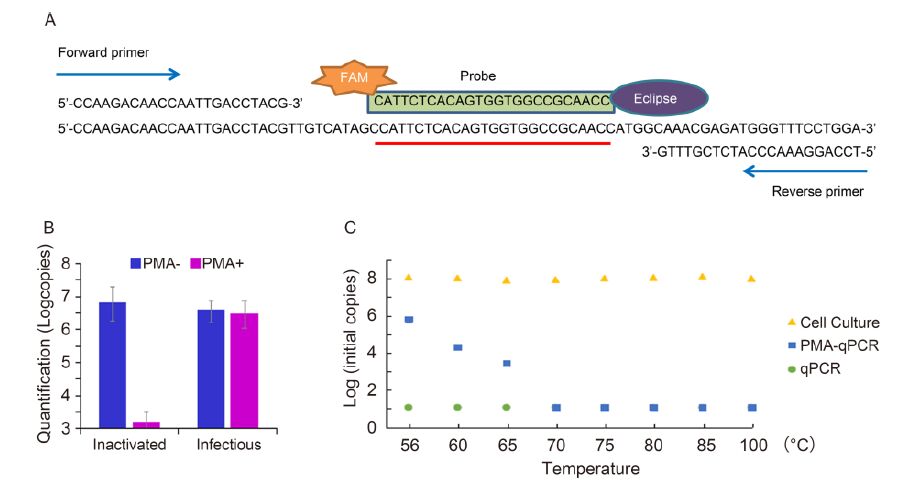
Figure 1. (A) Target fragment and primers. (B) Selective qPCR of viral suspensions (n = 3) with or without pretreatment with propidium monoazide (PMA). Dengue virus serotype 2 (DENV2) was inactivated by heating at 100 ℃. Titers were calculated using a standard curve generated by the amplification of a plasmid containing the target fragment. (C) Determination of infectious titer using cell culture and qPCR in DENV2 suspensions heated for 30 min at different tempera-tures. Selective qPCR indicates complete inactivation at 70 ℃.
For pretreatment with PMA, a 20 mmol/L stock solution was prepared by dissolving 1 mg of PMA (Biotium, Inc., Hayward, USA) in 98 µL of distilled water (dH2O), and the stock was stored at -20 ℃ until it was needed. The viral preparations were incubated in 50 µmol/L PMA for 5 min in the dark at 20 ℃, with occasional mixing. Then, the samples were crosslinked by exposing them to blue light-emitting diode (LED) light (Led-Active Blue, Philips, Holland) for 15 min on ice. The RNA was extracted using Buffer AVL-carrier RNA (Qiagen, Dusseldorf, Germany) according to the manufacturer's instructions, and then reverse transcription was performed at 37 ℃ for 15 min, using the PrimeScript™ RT reagent kit (TAKARA Co., Dalian, China). The reaction was terminated by incubation at 85 ℃ for 5 s. The samples were amplified in a Mx3005P Real-time PCR system (Stratagene, California, USA) with the following schedule: over 40 cycles of denaturation at 95 ℃, annealing at 60 ℃, and then elongation at 72 ℃. The total reaction mixture volumes were 25 µL and consisted of 1 µL of Premix Ex Taq (TAKARA, Co., Dalian, China), 0.5 µL of each of the forward and reverse primers, 0.5 µL of TapMan probe, 0.5 µL of ROX Ⅱ, 12.5 µL of cDNA, and 9.5 µL of double dH2O (ddH2O).
To generate the particles, 140-µL aliquots of the DENV2 stock were heated at 100 ℃ for 10 min, and then the target fragments from both thermal inactivation viral particles and infectious viral particles (7×103 PFU/mL) with or without pretreatment with PMA (Figure 1B) were amplified equally and efficiently. However, the compound significantly inhibited (P < 0.05) the amplifi-cation of the non-infectious viruses. It has been reported that high temperatures can inactivate numerous viruses, resulting in the loss of viral infectivity. Indeed, PMA has been demonstrated to prevent PCR-based detection of inactivated viruses or bacteria, but not of infectious orga-nisms (Nocker et al., 2009; Fittipaldi et al., 2010).
In addition, the ability of the cell cultures and selec-tive qPCR to measure the infectious titers was evaluated by using DENV2 suspensions heated for 30 min at 56 ℃, 60 ℃, 65 ℃, 70 ℃, 75 ℃, 80 ℃, 85 ℃, and 100 ℃. The qPCR performed without pretreatment with PMA detected 107.84 to 108.04 copies/µL of DENV2, although the infectious particles were not detected by cell culture (Figure 1C). However, the infectious titer decreased li-nearly at the inactivation temperature when the samples were pretreated with PMA. Indeed, the infectious particles were undetectable in the samples heated above 70 ℃, indicating their complete inactivation.
Serum samples of patients diagnosed with dengue fever were collected during the acute phase of the disease. qPCR with probes designed against all four dengue serotypes was performed for these samples within 1 h of the blood draws, to measure the viral titer. DENV2 was detected in two samples at 107.48 and 107.90 copies/µL. However, the titers were lower at 107.01 and 106.89 copies/µL, when the samples were pretreated with PMA, indicating that inactivated viral particles were present (Table 1).

Table 1. Application of propidium monoazide (PMA)-qPCR to detect clinical samplesa
The gold standard for measuring viral titers is titration in cell cultures, which is time-consuming. Furthermore, some viruses that pose significant public health concern are either difficult to culture or are nonculturable (Pecson et al., 2009). For instance, cell culture systems for noroviruses have not been established (Wigginton and Kohn, 2012). On the other hand, viruses such as the hepa-tits A virus and rotaviruses are not cytopathic in buffalo green monkey kidney cells (Parshionikar et al., 2010). An alternative approach is to quantify the viral genomes by qPCR, which cannot assess infection risk because it measures both infectious and non-infectious particles (Fittipaldi et al., 2010). Therefore, it is necessary to pretreat samples with PMA to selectively amplify the viable organisms.
The gold standard for measuring viral titers is titration in cell cultures, which is time-consuming. Furthermore, some viruses that pose significant public health concern are either difficult to culture or are nonculturable (Pecson et al., 2009). For instance, cell culture systems for noroviruses have not been established (Wigginton and Kohn, 2012). On the other hand, viruses such as the hepa-tits A virus and rotaviruses are not cytopathic in buffalo green monkey kidney cells (Parshionikar et al., 2010). An alternative approach is to quantify the viral genomes by qPCR, which cannot assess infection risk because it measures both infectious and non-infectious particles (Fittipaldi et al., 2010). Therefore, it is necessary to pretreat samples with PMA to selectively amplify the viable organisms.
The result of this assay indicates that the envelopes and capsids are catastrophically lost only above 70 ℃, while the infectivity is probably too low to induce obvious cytopathic effects when DENV2 suspensions are heated at 56 ℃-65 ℃. Therefore, the PMA-qPCR produces weakly positive results, while the cell culture method produces negative results. It is also conceivable that a virus may lose its infectivity although the capsid remains intact, such as when it loses the ability to bind host cell receptors (Fittipaldi et al., 2010). This indicates that the selective qPCR may provide some information on the extent of damage to viral envelopes and capsids, therefore, may be appropriate for assessing disinfection practices. Furthermore, this study established a relationship between the temperature of the process condition and stability of intact DENV2 particles, which could contribute to the design of studies to investigate the features of DENV2.
In summary, to the best of our knowledge, this is the first report of a technique that involves pretreating samples with PMA to greatly enhance the specificity for infectious particles detection. For example, the nonselective PCR of the sera from convalescent patients may still produce positive results because of the presence of inacti-vated or non-encapsulated particles. Moreover, the posi-tive results obtained by selective qPCR indicate that vi-ruses continue to replicate and, therefore, the infection may progress further. While these results are clinically significant, additional clinical samples of acute phase serum would be required to provide valuable information on the accuracy of the PMA-qPCR method. The technique, qPCR combined with PMA pretreatment, is expected to be applied in the diagnosis of dengue infection. Factors that may affect this approach should be investigated in detail in future research.
-
This study was partly supported by the Joint Funds of NSFC-Guangdong of China under Grant (No.U1132002), by the National Natural Science Foundation of China (No. 31270974, 31470271), and by the Technologies R & D Program of Guangdong Province (No. 2013A020229004) and Guangzhou city (No. 201508020263). The authors declare that they have no conflict of interest. All partic-ipants provided written informed consent. The whole study was approved by ethics committee of Southern Medical University, China.
Supplementary figure/table are available on the websites of Virol-ogica Sinica: www.virosin.org; link.springer.com/journal/12250
-
Figure S1. The amplification plots (A) and standard curve (B) constructed by serial 10-fold dilutions of the standard plasmid pMD-DENV2
Probe/Primer Sequence (5'-3') Forward primer CCAAGACAACCAATTGACCTACG Reverse primer TCCAGGAAACCCATCTCGTTTG Probe FAM-CATTCTCACAGTGGTGGCCGCAACC-Eclipse Target fragment CCAAGACAACCAATTGACCTACGTTGTCATAGCCATTCTCACAGTGGTGGCCGCAACCATGGCAAACGAGATGGGTTTCCTGGA Table S1. Primers and probe

 DownLoad:
DownLoad: