










-
Dear Editor,
Human papillomaviruses (HPV) are a large group ( > 200 genotypes) of small double-stranded DNA viruses (https://pave.niaid.nih.gov/). Although infections by most HPV types are asymptomatic, persistent infections in cervical and ano-genital epithelia by high-risk HPV can lead to the development of cervical and ano-genital intraepithelial neoplasia and invasive carcinoma. Of 15 known high-risk HPV types, HPV16 and HPV18 are the two most common types inducing the development of cervical, anal, and a small portion of oropharyngeal cancers and are found in~70% of cervical cancer cases worldwide (Munoz et al., 2003).
Active HPV18 DNA amplification depends on keratinocyte differentiation and requires viral DNA helicase E1 and E2, an accessory factor for E1 binding to the viral replication origin (Ori) (Demeret et al., 1995). We recently constructed a full transcription map of HPV18 from raft cultures with productive HPV18 infection and identified two major transcription start sites (TSS), one at nt 55 (TSS-55) and the other at nt 102 (TSS-102), for transcription of viral early transcripts (Wang et al., 2011). Analyses of the region 5′ to each TSS showed a TATA box (a eukaryotic core promoter motif) 27 bp upstream of the TSS-55 and 25 bp upstream of the TSS-102 (Figure 1A). We designated the sequence upstream of each TSS, respectively, as the promoter P55 and the promoter P102 (Wang et al., 2011). Interestingly, the P55 promoter is positioned in a core Ori region (Demeret et al., 1995) which contains three E2 binding sites (Figure 1A). The identified TATA box for the promoter P55 is within the A+T-rich region of the core Ori, whereas the TATA box for the promoter P102 is outside of the core Ori (Demeret et al., 1995; Demeret et al., 1998). Thus, we are assuming that the viral DNA replication might prevent the transcription of P55 promoter. Using poly A+ total RNA isolated from either 8-day-old or day-16-old rafts derived from HPV18-infected human foreskin keratinocytes (HFK18) for TSS assays by 5′-rapid amplification of cDNA ends (5′ RACE), a powerful method to specifically amplify the 5′ end of a given transcript (Wang et al., 2011; Wang and Zheng, 2016), we discovered that RNA transcription from both P55 and P102 promoters were equally active in the 8-day-old rafts, but this transcription from the P55 promoter faded in the 16-day-old rafts where the P102 promoter activity remained unchanged (Figure 1B). It has been demonstrated that active HPV18 DNA replication is associated with keratinocyte differentiation and becomes robust after day 10 in the raft culture (Wang et al., 2009). To further correlate the cell differentiation to the blockade of P55 promoter activation, we examine human foreskin keratinocytes immortalized with HPV18 infection (HFK18 cells) (Meyers et al., 1997; McLaughlin-Drubin and Meyers, 2005) either in the low calcium or in high calcium condition for transcription activities of both P55 promoter and P102 promoter by 5′ RACE (Wang et al., 2011; Wang and Zheng, 2016). Calcium has been routinely used to induce keratinocyte differentiation in HPV field (Jones et al., 1997; Moody and Laimins, 2009). Under high calcium condition, a notable reduction of the transcription activity was found for the P55 promoter, but not for the P102 promoter in the HFK18 cells (Figure 1C), similar to what we observed in HPV18-infected day-16-old rafts. These data suggest that human keratinocyte differentiation and HPV18 DNA replication inhibits RNA transcription from the P55 promoter effectively.
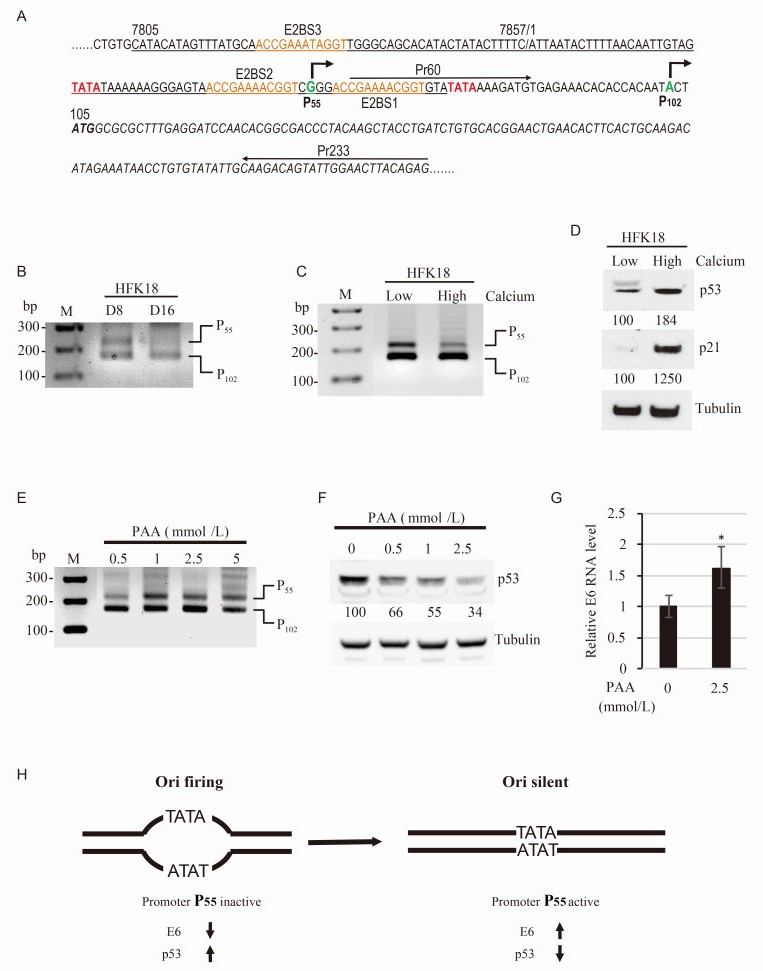
Figure 1. Viral DNA replication inactivates the transcription from HPV18 P55 promoter and prevents viral E6 expression
The promoter P102-derived early transcripts contain an extremely short 5′ untranslational region (UTR), with only three nucleotides upstream of the viral E6 open reading frame (ORF) in which its first translation initiation codon ATG starts at nt 105 position in the HPV18 genome (Figure 1A). This short 5′ UTR structure of the P102 transcripts would be impossible for recruiting ribosome binding to initiate E6 translation. In contrast, the promoter P55-derived RNA transcripts have a much longer 5′ UTR (51 nts) upstream of the E6 ORF (Figure 1A) and should have enough space for ribosome recruiting and binding to initiate E6 translation. Thus, the P55 promoter-derived RNA transcripts would be the RNA transcripts to translate E6 protein, although both transcripts derived from the P55 promoter and P102 promoter are capable for translation of E7 and other viral early proteins after alternative RNA splicing (Tang et al., 2006). If this assumption is true, the reduced transcription from the P55 promoter in the differentiated keratinocytes with active viral DNA replication would decrease E6 expression and lead to stabilize host p53, because viral E6 interacts with p53 and induces p53 degradation via E6-mediated ubiquitination (Scheffner et al., 1993). As expected, we found that the p53 level in the HFK18 cells was increased by 84% in the high calcium condition over that in the low calcium condition by Western blotting using an anti-p53 antibody (Ab-6, Calbiochem, San Diego, CA, USA), accompanied by 12-fold increase of p21, a p53-mediated transcription product (Figure 1D).
To determine if viral DNA replication directly inactivates the P55 promoter activity, we added phosphonoacetic acid (PAA), a chemical compound that inhibits DNA polymerases (Allaudeen and Bertino, 1978), to inhibit HPV18 DNA replication in calcium-differentiated HFK18 cells. As shown in Figure 1E and 1F, PAA at 1 mmol/L or above did induce a much higher level of RNA transcription from the P55 promoter, but had little effect on the P102 promoter (Figure 1E), accompanied by reduction of p53 protein level in a dose-dependent manner (Figure 1F), although PAA at 5 mmol/L might exhibit some toxicity to the cells. Consistent with the increased P55 promoter transcription and p53 level reduction, we saw a greater production of the P55 promoter-derived E6 RNA in the HFK18 cells treated with PAA (Figure 1G). Altogether, these data provide strong evidence that transcription of the HPV18 P55 promoter is directly regulated by viral DNA replication and is responsible for viral E6 production to modulate host p53 stability during virus infection.
HPV18 DNA replication requires an A+T-rich Ori in the long control region (LCR) and the characterized core Ori (Demeret et al., 1995; Demeret et al., 1998) contains the binding sites for two viral DNA-binding proteins, E1 as a DNA helicase and E2 as an accessory factor to E1 (Demeret et al., 1995; Kuo et al., 1994). In addition to its role in viral DNA replication, E2 has been described as a transcriptional activator or repressor to regulate viral early promoter activity through consensus E2-binding sites (Romanczuk et al., 1990), upstream of the viral early promoter. 5′ RACE is a powerful method to specifically amplify the 5′ end of a transcript to map the transcription start sites and efficiency of the individual promoter usage (Wang et al., 2011; Wang and Zheng, 2016). By using this method in this study, we demonstrated that HPV18 viral DNA replication inactivates RNA transcription from viral early promoter P55, but not the P102 promoter. Our data thereby provides the direct evidence that HPV18 DNA replication contributes to another layer of the control for viral E6 expression.
HTML
-
This study was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute and the Center for Cancer Research. The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: