











-
Porcine epidemic diarrhea virus (PEDV) is the etiological agent of porcine epidemic diarrhea (PED) with symptoms including diarrhea, vomiting, anorexia, and dehydration (Pensaert and de Bouck, 1978). PEDV causes significantly high mortality in piglets under 7 days of age though pigs of all ages can be infected (Chen et al, 2013; Vlasova et al, 2014). Since its identification in 1984 in China, PEDV has continuously caused problems to the pig farming industry, and the situation aggravated because of the emergence of highly pathogenic PEDV variants in China in 2010 (Xuan et al, 1984; Li et al, 2012; Zhang et al, 2013). Recently, outbreaks of infections by novel variant PEDV strains with recombination and heterogenic Spike (S) genes have been reported in several provinces of China with a positive rate of up to 72% (Chen et al, 2013; Cai et al., 2016; Li et al, 2016a; Wang et al, 2016).
PEDV is a member of coronavirus (CoV) belonging to the genus Alphacoronavirus (α-CoV) and has intestinal tropism (Pensaert and de Bouck, 1978; Xuan et al, 1984; Belouzard et al, 2012). CoVs are enveloped viruses of the family Coronaviridae and contain a positive, single-stranded RNA genome (Masters and Perlman, 2013). Until now, 4 genera of CoVs have been identified: α-CoV, Betacoronavirus (β-CoV), Gammacoronavirus (γ-CoV), and Deltacoronavirus (δ-CoV) (Perlman and Netland, 2009; Woo et al, 2012). Members of α-CoV and β-CoV infect a wide range of mammals including humans and lead to respiratory, gastrointestinal, hepatic, and neurological diseases (Perlman and Netland, 2009; Graham et al, 2013).
The spike protein of CoVs is a membrane glycoprotein that contains important antigenic determinants and neutralizing epitopes and plays a key role in mediating the entry of the CoVs into host cells through receptor binding and membrane fusion (Cruz et al, 2008; Sun et al, 2008; Li, 2015). Previous researches have shown that the S gene is one of the most variable genes in the PEDV genome and that it is a useful marker for determining the genetic diversity of PEDV (Chen et al, 2013; Tian et al, 2014). In recent years, PEDV has spread throughout the Hubei Province of China and adversely affected pig farming. In this study, to determine the infection status and to better understand the molecular characteristics of PEDV in different districts in Hubei, we screened PED samples for PEDV and investigated the full-length S genes. Our results revealed the coexistence of multiple PEDV genotypes with novel genetic mutant sites in the S genes in Hubei. These findings can help better understand the epidemic status of PEDV as well as aid in developing new vaccines for the control and prevention of PED.
-
During 2016, there were PED outbreaks in 34 farms distributed in 6 districts in Hubei province (Figure 1). The clinical features were characterized by watery diarrhea, acute vomiting, anorexia, and dehydration. A total of 172 porcine samples including intestinal, fecal swab, and feces samples were collected from all 34 farms and transported on ice to the Hubei Animal Disease Prevention and Control Center.
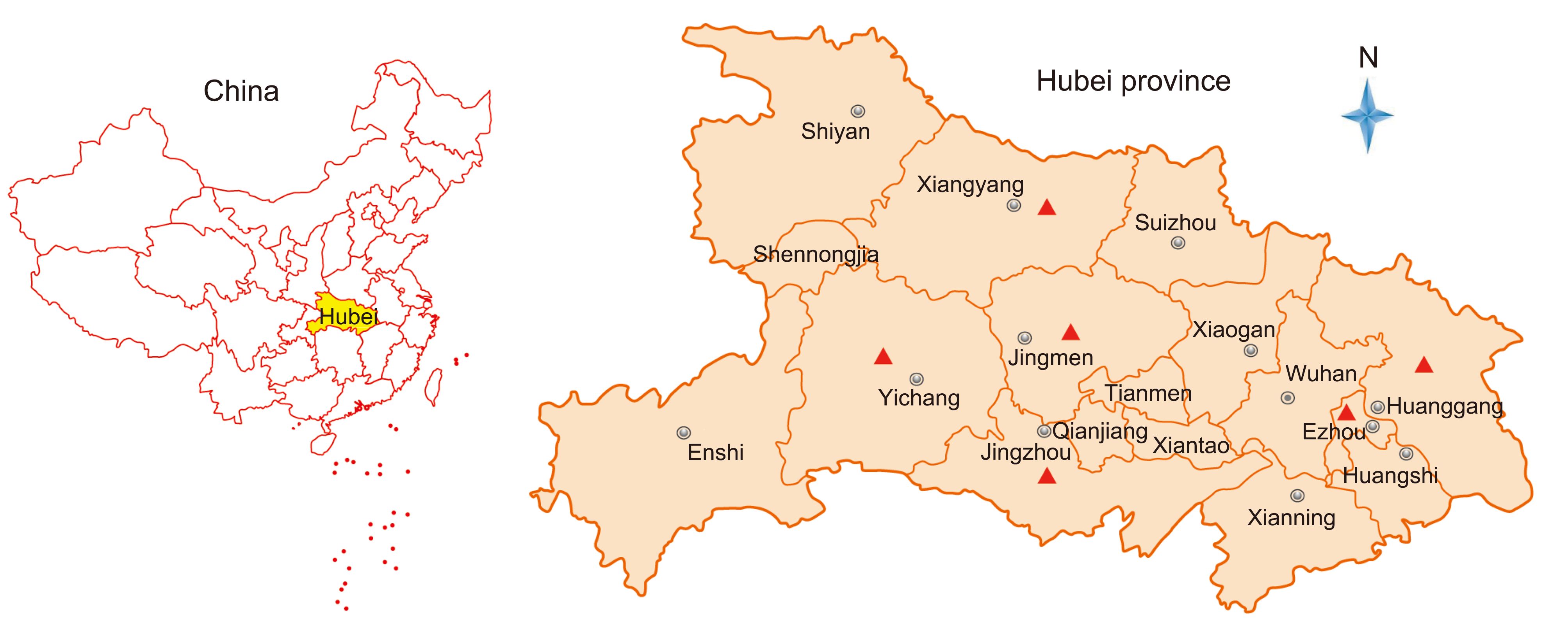
Figure 1. Map of sampling sites in the Hubei Province of China. Red triangles indicate the six districts from where samples were collected.
-
The fecal swab and feces samples were vortexed for 2 min and centrifuged at 3000g for 5 min. For the intestinal samples, 1 g of the tissue was homogenized in 10 mL of phosphate buffer saline (PBS) and centrifuged at 3000g for 10 min. Viral RNA was extracted from 140 μL of the supernatants using the QIAamp Viral RNA Mini Kit (Qiagen) following the manufacturer’s instructions and eluted in 60 μL buffer AVE (Qiagen). PEDV RNA was detected using RT-PCR targeting the conserved regions of the PEDV nucleocapsid (N) protein gene as described by Kim et al. (2007), with modifications to fit the ABI StepOne system. Briefly, using the TaqMan One-Step RT-PCR Kit (Applied Biosystems), RT-PCR was performed in a 25 μL reaction mix containing 4 μL RNA, 1× RT-PCR enzyme mix, 1× RT-PCR buffer, 40 pmol forward primer (5′-CGCAAAGACTGAACCCACTAATTT-3′), 40 pmol reverse primer (5′-TTGCCTCTGTTGTTACTTGGAGAT-3′) and 12 pmol probe (Cy5-TGTTGCCATTGCCACGACTCCTGC-BHQ3). Amplification parameters were as follows: an initial step of 10 min at 50 °C, 10 min at 95 °C, followed by 45 cycles of 15 s at 95 °C and 20 s at 60 °C.
-
The samples positive for PEDV in the RT-PCR were then selected to amplify the S genes. By aligning the genomes of representative PEDV strains, a pair of primers was designed to amplify complete S genes: primer SF (5′-GCAAGTGGCGCTGTGATTGACG-3′) and SR (5′-AGACTTCGAGACATCTTTGACA-3′) corresponding to the locations of 20378-20399 and 24827-24848 nt in the PEDV genomes (reference: AF353511). PCR amplification was performed in a 25 μL reaction mixture using One-step RT-PCR (Invitrogen Platinum SuperScript III, Invitrogen, San Diego, USA) under the following parameters: 5 min at 90 °C, 35 cycles of 30 s at 94 °C, 30 s at 55 °C, and 4.5 min at 68 °C, followed by a final 10-min extension at 72 °C. The PCR products were then gel-purified and sequenced on the 3100 Sequencer using SF and SR and 5 other inner sequencing primers. Weak bands that could not be sequenced directly were cloned into pGEM-T Easy Vector (Promega, WI, USA). Positive plasmids containing the insertion of the S genes were verified by colony PCR. For each sample, at least 5 positive colonies were sequenced. Standard precautions were taken to avoid PCR contamination and no false positives were observed in the negative controls.
-
The obtained sequences for each sample were assembled to contigs covering the complete S genes using assembly software implemented in Geneious package (Version 10.0.7) (Kearse et al, 2012). Contigs were both-end trimmed to obtain the complete ORF coding regions of the S gene using BioEdit (Version 7.1.9). Sequence alignment was performed using Translation Align in Geneious, and phylogenetic trees were constructed using the maximum likelihood (ML) method with bootstrap values determined by 1000 replicates in MEGA6 and PhyML software (Criscuolo, 2011; Tamura et al, 2013).
-
During 2016, PED outbreaks had continuously occurred in multiple districts of the Hubei Province in China, even though most of the farms had been vaccinated using the bivalent attenuated TGEV and PEDV vaccine. In total, 172 clinical samples covering all ages of swine, including fecal swab, intestinal, and feces samples, were collected from 34 farms with different scales (from 100–15000) located in 6 districts in the Hubei province of China (Figure 1, Table 1, Supplementary Material S1).
Location Farm numbers Sample types PEDV positives/total Total positives Ezhou 6 Feces 8/31 8/31(25.8%) Huanggang 3 Feces 24/25 28/29(96.6%) Intestine 4/4 Xiangyang 8 Feces 0/8 14/40(35%) Fecal Swab 1/16 Intestine 13/16 Jingzhou 9 Feces 19/29 19/29(65.5%) Yichang 5 Feces 1/29 1/29(3.4%) Jingmen 3 Feces 0/4 4/14(28.6%) Fecal Swab 4/10 Total 34 – – 74/172(43%) Table 1. PEDV-positive samples among those collected from 6 districts in the Hubei province of China
By RT-PCR detection of the N gene, of the 172 porcine samples, 74 samples (43%) were found to be PEDV-positive (Table 1, Supplementary Material S1). Among the 34 farms, 20 (58.8%) were positive for PEDV, but different districts showed different positive rates ranging from 3.4% (Yichang) to 96.9% (Huanggang) (Table 1, Supplementary Material S1).
-
To investigate the genetic diversity and molecular epidemiology of PEDV in different districts in the Hubei Province of China, the complete S genes of the 74 clinical isolates were amplified and sequenced. Comparison of the nucleotide sequence (nt) with those of the PEDV S genes in GenBank revealed that the complete S genes of the 74 isolates encodes 21 different spike proteins with amino acids (aa) mutations (Supplementary Material S2). The sequences obtained were named using the abbreviation of district origin plus the serial number. The 21 representative S gene sequences were deposited in GenBank under the accession numbers KY775035–KY775055. The overall nt and aa identities between theseS genes and the previously reported S genes ranged from 92.7% to 99.9% and 91.4% to 98.8%, respectively (Supplementary Material S3). Additionally, these 21 sequences had 4 different lengths: 9 sequences (HBXY-1, HBXY-2, HBXY-4, HBXY-5, HBJZ-1, HBJZ-2, HBEZ-1, HBJM-3, and HBHG-6) had 4161 nt encoding 1386 aa; 8 sequences (HBJM-1, HBJM-2, HBXY-3, HBHG-1, HBHG-2, HBHG-3, HBHG-4, and HBHG-5) had 4158 nt encoding 1385 aa; 3 sequences (HBEZ-2, HBEZ-3, and HBEZ-4) had 4152 nt encoding 1383 aa; and 1 sequence (HBYC-1) had 4149 nt encoding 1382 aa (Figure 2, Supplementary Material S3, S4, S5).

Figure 2. Phylogenetic analysis of full-length amino acid sequences of spike proteins of PEDV. The phylogenetic tree was constructed based on the maximum likelihood method using a Poisson model under 1000 replicates of bootstrap values; for each node, bootstraps ≥ 50% are shown. The scale bar represents 0.005 substitutions per amino acid. The strain names, isolation years and places, and GenBank accession numbers are shown. S (nt) and S (aa) indicate the complete length of the nucleotide and amino acid sequences of the S genes and S proteins, respectively. The results for the GI, GII, and INDEX-like genogroups were in accordance with previous studies; subgroups shown in the figure were proposed in this study for better description of the genetic diversity of spike proteins. Spike sequences detected in this study are colored and in bold.
With regard to insertions and deletions, the 21 S proteins could be divided into 2 types. Seventeen Type I sequences showed the same features: 2 insertions of 4 aa (56GENQ59 or 56GENH59) and 1 aa (140N or 140D) and a deletion of 2 aa (160GK161). Nine of them (HBJM-1, HBJM-2, HBHG-1, HBHG-2, HBHG-3, HBHG-4, HBHG-5, HBXY-3, and HBYC-1) had a deletion of H at position 1197. Four Type II sequences did not show the above deletions and insertions (Figure 3). We did not find any new deletion or insertion sites in these S genes.
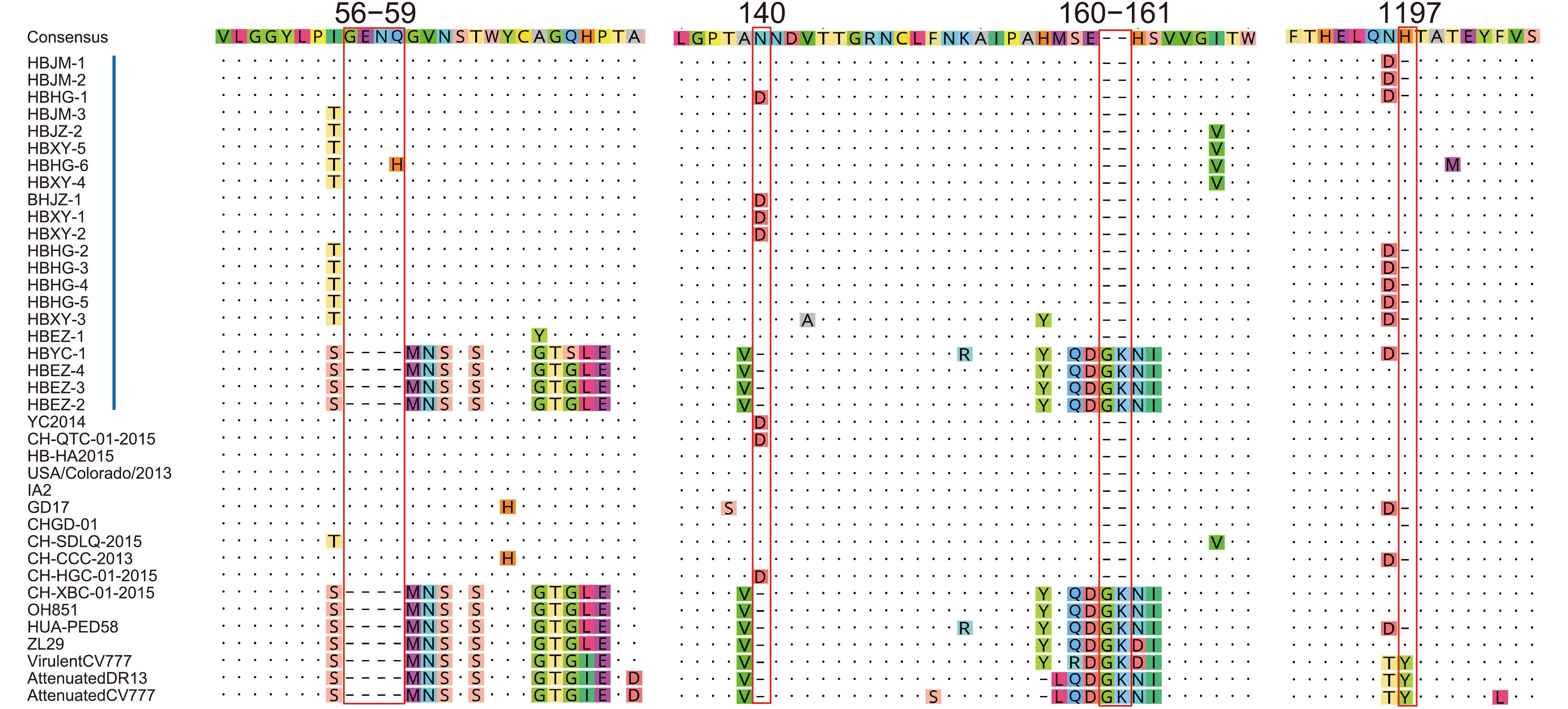
Figure 3. Insertions and deletions in the spike proteins of PEDV. Red bars indicate the precise positions of 4 insertions and deletions in the spike proteins. Vertical blue bars indicate the strains in this study. The reference strain names are shown, and their GenBank accession numbers are the same as those indicated in Figure 2.
-
The phylogenetic tree was constructed based on the aa sequences of the spike proteins encoded by the complete S genes along with those of previously reported PEDV isolates (Supplementary Material S5). In the tree, of the 21 sequences, 17 were dispersed into 5 subgroups in the GII group, one (HBYC-1) was clustered into the GI group but formed a new subgroup, and 3 were grouped with the INDEX-like strains (Figure 2). For more precise analysis, phylogenetic analysis of full-length aa se- quences of the spike proteins of PEDV and transmissible gastroenteritis virus (TGEV) were constructed. The TGEV was used as an out-group (ABG89301). The result showed that there were great varieties between S gene of PEDV and S gene of TEGV and a long evolutionary branch between TEGV and PEDV was observed based on the S tree (Supplementary Material S6). The topologies of the trees with or without TGEV were similar, but the tree without TGEV can better display the differences among PEDV strains (Figure 2).
The GII-1 subgroup consisted of HBXY-1, HBXY-2, and HBJZ-1 and isolates from other Chinese provinces (Jiangsu, Henan, Guangdong, Zhejiang, etc.). To the best of our knowledge, this is the first report of GII-1 subgroup detection in Hubei. The aa sequences of HBXY-1, HBXY-2, and HBJZ-1 showed >95% identities with those of other isolates. The GII-2 subgroup consisted of HBEZ-1 and another isolate that was also from Hubei (HB-HA2015) with 98.7% aa identities. The GII-3 subgroup comprised HBJM-1, HBJM-2, and HBHG-1 and a GD17 isolate previously reported from the Guangdong Province of China; the identities between HBJM-1, HBJM-2, HBHG, and GD17 ranged from 98.4% to 98.6% at the aa level. The GII-4 subgroup comprised HBXY-3, HBHG-2, HBHG-3, HBHG-4, and HBHG-5 and isolates previously reported from Henan, Guangdong, Guangxi, Yunnan, and other provinces of China. The 5 sequences were most similar to the sequence of the CH-CCC-2013 strain, with 97.8% to 98.5% aa identities. The GII-5 subgroup comprised HBJM-3, HBHG-6, HBXY-4, HBJZ-2, and HBXY-5 and CH-SDLQ, which is an isolate reported from the Shandong Province of China in 2015. These 5 sequences showed 98.9% to 99.4% aa identities with the sequence of CH-SDLQ.
The GI-1 subgroup consisted of two strains, HBYC-1 and HUA-PED58. HUA-PED58 is a novel strain detected in Vietnam in 2015. These 2 strains had 99.1% aa identities. In the phylogenetic tree, HBYC-1 and HUA-PED58 formed a distinct subgroup in the GI group, which indicated that these two strains were genetically distanced to typical GI strains.
The last INDEX-like subgroup consisted of HBEZ-2, HBEZ-3, and HBEZ-4 and other isolates previously reported from USA, Germany, and China. In 2014, a variant PEDV OH851 strain was found in USA. The S gene of OH851 has similar insertions and deletions as the prototype PEDV discovered in 2013. In 2015, an INDEX-like PEDV was found in the Hubei (ZL29 strain) and Guangdong (CH-XBC) provinces of China. The S genes of HBEZ-2, HBEZ-3, and HBEZ-4 were closest to that of CH-XBC, with 99.5%–99.7% aa identities.
-
The analyses of the S gene sequences revealed that 7 different PEDV genotypes were circulating in Hubei in 2016. These PEDV strains not only covered all the typical genotypes of PEDV, including GI and GII, but also contained the INDEX type and some novel genotypes such as HBYC-1 in the GI-1 subgroup. In addition, the results also showed that several genotypes of PEDV were epidemic in one district: for example, GII-1, GII-4, and GII-5 strains coexisted in the Xiangyang district; GII-1 and GII-5 strains coexisted in the Jingzhou district; GII-3, GII-4, and GII-5 strains coexisted in the Huanggang district; and GII-2 and INDEX strains were both found in the Enshi district.
-
The S protein of PEDV is a membrane glycoprotein and contains 4 major important epitopes (Cruz et al, 2008; Sun et al, 2008). We compared these 4 major epitopes and found that, between the Hubei 2016 PEDV strains and other earlier isolates, there were no aa mutations in the epitope 753YSNIGVCK760. All the spike proteins of the Hubei 2016 PEDV strains contained the 769SQSGQVKI776 epitope but not 769LQDGQVKI776; the conserved 1373GPRLQPY1379 epitope was found in all the spike proteins except HBEZ-4, which had 1373GPRLHPY1379 with a substitution of Q to H at aa position 1377. In the longest epitope at aa positions 504-643, 7 novel aa mutations were detected: the H to Y substitution at aa position 526 in HBXY-2; S to Y substitution at aa position 567 in HBXY- 1; F to L substitution at aa position 622 in HBHG-6; P to S substitution at aa position 636 in HBHG-1; E to G substitution at aa position 638 in HBXY-4; and T to K substitution at aa position 641 in HBHG-2, HBHG-3, HBHG-4, and HBHG-5 (Figure 4).
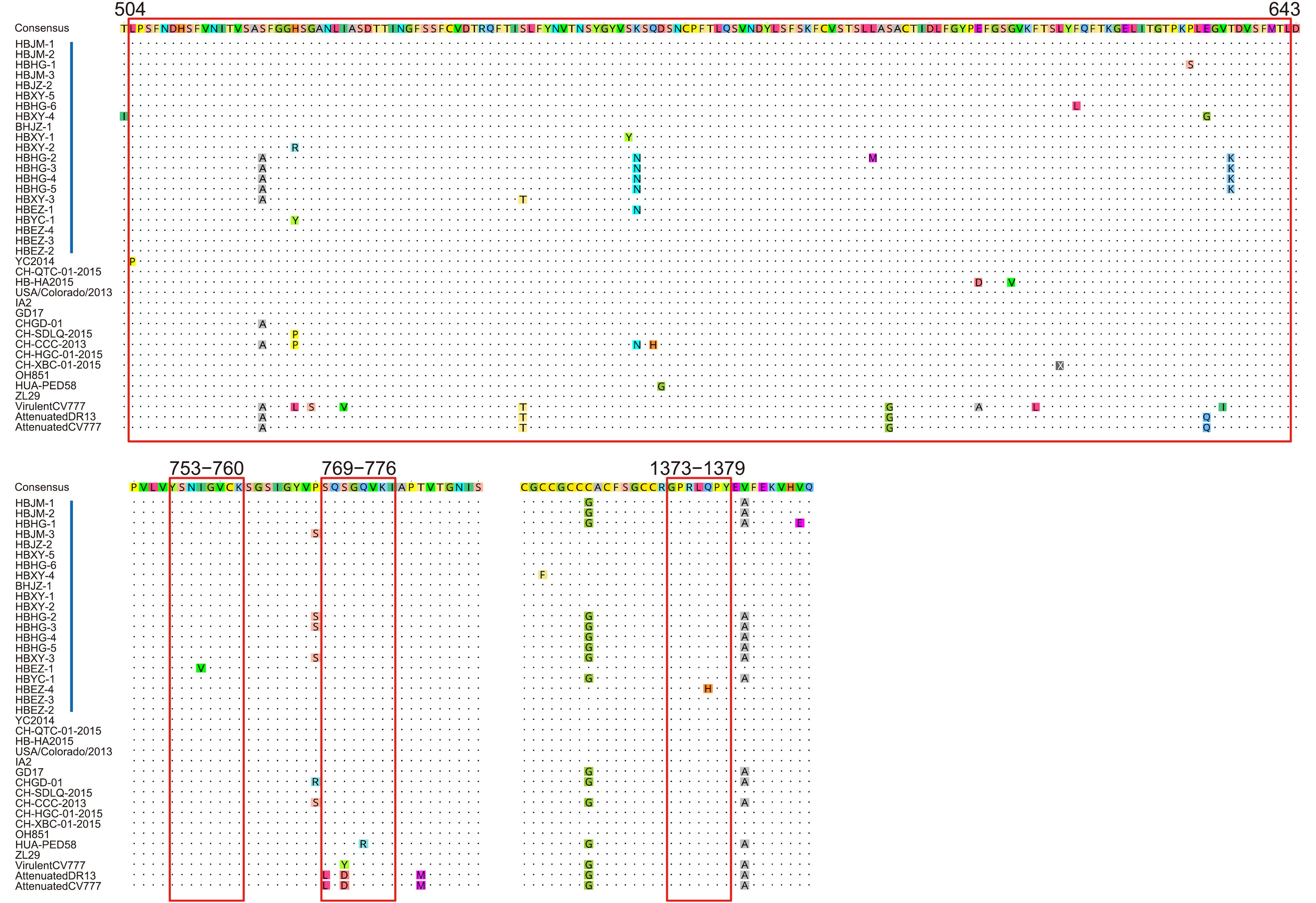
Figure 4. Antigenic variation in the spike protein of PEDV. Blue bars indicate the precise positions of 4 epitope regions. Vertical red bars indicate the strains in this study. Other labels and instructions are the same as those in Figure 3.
-
The spike protein of CoVs is responsible for cell receptor attachment and membrane fusion to mediate virus entry (Li, 2015). The S1 (residues 21-793) in the PEDV spike protein can bind aminopeptidase N (APN) as a receptor and N-acetylneuraminic acid (Neu5Ac) as a co-receptor (Huan et al, 2015; Liu et al, 2015). Recent studies have shown that the S 477_629 fragment located in the CTD (residues 253-638) and the S 1_320 fragment located in the NTD (residues 21-324) are essential for binding to APN and sugar, respectively (Deng et al, 2016). In this study, spike proteins were compared to elucidate the aa site changes. The results showed that, except HBJZ-1 and HBJZ-2, all the other 19 spikes had 2-11 new aa mutant sites in comparison with the reference sequences. Consistent with previous reports, more mutation sites were detected in S1 (44 sites) than in S2 (14 sites) (Supplementary Material S2). In addition, 23 aa changes were observed in the S 1_320 fragment, which is much higher than the 6 aa changes found in the S 477_629 fragment (Supplementary Material S2). Because the precise sites for receptor binding have not been characterized yet, whether these aa changes alter the capacity of receptor binding or have impacts on other characteristics of the virus, such as virulence, still needs to be explored in future studies.
-
PEDV epidemics jeopardize pig farming worldwide and more so in China, which holds a large number of pig farms. Since the emergence of new PEDV strains in China in 2010, PEDV has continued to cause economic losses in almost all districts in the Hubei Province of China despite several control methods including biosecurity and vaccination. One important reason for this is the emergence of novel PEDV variants (Jung and Saif, 2015; Li et al, 2016a; Li et al, 2016b). However, genetic information on PEDV in Hubei is quite limited; therefore, for better prevention and control of the spread of PEDV in the future, it is crucial to investigate the genetic diversity and variation of PEDV in fields.
By collecting clinical samples from pigs with PED in different districts in Hubei and by screening for PEDV, we confirmed 74 samples positive for PEDV among 172 samples (43%) from 34 farms distributed in 6 districts (Supplementary Material S1). However, we found that, of the 34 farms, 14 farms were free of PEDV, which suggested that the PED in these farms may be caused by other factors such as other viral pathogens commonly found in samples of enteric viral infections, including transmissible gastroenteritis virus (TGEV), astrovirus, and rotavirus (Zhang et al, 2013; Brnic et al, 2014; Molinari et al, 2016).
The gene encoding the spike protein of CoVs is an important gene responsible for viral entry and therefore has been extensively studied (Sun et al., 2008; Belouzard et al, 2012; Li et al, 2012; Wang et al, 2014; Li, 2015; Liu et al, 2015). The S gene of PEDV has been used widely as a molecular marker in studies on the genetic diversity of PEDV (Chen et al., 2013; Tian et al., 2014). Upon characterization of the complete S gene, we confirmed that 7 different genotypes of PEDV coexisted in the Hubei province in 2016. Based on genetic characteristics and emerging periods, PEDV can be sorted into 3 genogroups: GI (classical), GII (variant), and INDEL-variant group (Vlasova et al, 2014; Lee, 2015; Wang et al, 2016). Most of the PEDV strains in this study belonged to the GII group, which was consistent with the PEDV situation in China, where most of the highly prevalent variant PEDV strains in China have been found to be members of the GII genogroup since 2010 (Wang et al, 2016). However, new variant PEDV strains of the GI and INDEX groups were also found in this study. These results indicated that the epidemic heterogeneity of PEDV in Hubei is more complex than previously assumed.
Although we could not detect new recombinant events among the S genes, the coexistence of diverse PEDV genotypes in the same location may foster viral genomic recombination and promote the emergence of more novel PEDV strains (Tian et al, 2014; Li et al, 2016a). More worthy of concern is the fact we found 58 new aa mutant sites in the S genes in these field strains. In some districts, multivariants and accumulating aa mutations in the S gene could be observed in a herd. For example, we detected 3 different S variants in a herd in Ezhou: HBEZ-2, HBEZ-3, and HBEZ-4 (Figure 2, Supplementary Material S2). HBEZ-2 had 2 aa mutant sites (Y307D and H1047Q), HBEZ-3 had 3 aa mutant sites (L300S, Y307D, and H1047Q), and HBEZ-4 had 4 aa mutant sites (L300S, Y307D, H1047Q, and H1377Q).
Whether these mutants can lead to higher virulence or escape from immunoprotection from classical vaccines is unknown. However, it is important to reassess the efficacy of the current PEDV vaccines against the emerging variant strains to determine cross-protection. Further isolation and sequencing of full-length genomes of these PEDV strains will be helpful in determining their pathogenicity and subtle genetic characteristics. Living PEDV strains will be useful for assessing the protective efficacy of current vaccines and could be used for developing new vaccines for PEDV infections.
-
This work funded by the National Natural Science Foundation of China (31470260 and 81401672) and the "Fundamental Research Funds for the Central Universities" (531107040975).
-
The authors declare that they have no conflict of interest. The whole study was approved by the Hubei Animal Disease Prevention and Control Center (Wuhan, Hubei, China). All institutional and national guidelines for the care and use of animals were followed.
-
XYG, GQP, and TTL designed the experiment. NHS, FHP, TTL, and XJ collected samples. ZZ and TTL carried out the most of the experiments. XYG, GQP, TTL, and ZZ analyzed the data. XYG checked and finalized the manuscript. All authors read and approved the final manuscript.
Supplementary materials are available on the websites of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-

Table S1. PEDV-positive samples among those collected from 34 farms in 6 districts in the Hubei Province of China

Table S2. New amino acid mutations in the spike proteins of field strains in Hubei in 2016
S3. Nt and aa pairwise identities of S genes and the encoded spike proteins
S4. A fasta format file of the nt pairwise alignment of S genes
S5. A fasta format file of the aa pairwise alignment of spike proteins

Figure S6. Phylogenetic analysis of full-length amino acid sequences of spike proteins of PEDV and transmissible gastroenteritis virus (TGEV). The phylogenetic tree was constructed based on the maximum likelihood method using a Poisson model under 1000 replicates of bootstrap values; for each node, bootstraps ≥ 50% are shown. The scale bar represents 0.005 substitutions per amino acid. The strain names, isolation years and places, and GenBank accession numbers are shown. S (nt) and S (aa) indicate the complete length of the nucleotide and amino acid sequences of the S genes and S proteins, respectively. The results for the GI, GII, and INDEX-like genogroups were inconsistent with those of previous studies; subgroups shown in the figure were proposed in this study for better description of the genetic diversity of spike proteins. Spike sequences detected in this study are colored and in bold. The TGEV was used as an out-group
Coexistence of multiple genotypes of porcine epidemic diarrhea virus with novel mutant S genes in the Hubei Province of China in 2016
- Zhe Zeng 1,# ,
- Ting-Ting Li 2,# ,
- Xin Jin 2 ,
- Fu-Hu Peng 2 ,
- Nian-Hua Song 2 ,
-
Gui-Qing Peng
1,,
 ,
, -
Xing-Yi Ge
3,,
- Received Date: 23 May 2017
- Accepted Date: 05 July 2017
- Published Date: 26 July 2017
Abstract: The emergence of highly virulent porcine epidemic diarrhea virus (PEDV) variants in China caused huge economic losses in 2010.Since then,large-scale sporadic outbreaks of PED caused by PEDV variants have occasionally occurred in China.However,the molecular diversity and epidemiology of PEDV in different provinces has not been completely understood.To determine the molecular diversity of PEDV in the Hubei Province of China,we collected 172 PED samples from 34 farms across the province in 2016 and performed reverse transcription polymerase chain reaction (RTPCR) by targeting the nucleocapsid (N) gene.Seventy-four samples were found to be PEDVpositive.We further characterized the complete spike (S) glycoprotein genes from the positive samples and found 21 different S genes with amino acid mutations.The PEDV isolates here presented most of the genotypes which were found previously in field isolates in East and SouthEast Asia,North America,and Europe.Besides the typical Genotypes I and II,the INDEX groups were also found.Importantly,58 new amino acids mutant sites in the S genes,including 44 sites in S1 and 14 sites in S2,were first described.Our results revealed that the S genes of PEDV showed variation and that diverse genotypes of PEDV coexisted and were responsible for the PED outbreaks in Hubei in 2016.This work highlighted the complexity of the epidemiology of PEDV and emphasized the need for reassessing the efficacy of classic PEDV vaccines against emerging variant strains and developing new vaccines to facilitate the prevention and control of PEDV in fields.

 DownLoad:
DownLoad: