









-
The chikungunya virus (CHIKV) is a member of the genus Alphavirus and is transmitted through the bites of infected Aedes mosquitoes (Tsetsarkin et al., 2011a). Since November 2016, more than 4,000 cases of chikungunya fever cases have been reported in Pakistan (Rauf et al., 2017). The total number of people affected by the chikungunya outbreak is likely to be more than 30,000, based on the estimations of different healthcare authorities (Rauf et al., 2017). CHIKV was first described in Africa in 1952 and has been found to circulate in more than 60 countries in Africa, Asia, Europe, and Americas in the following decades (Ng and Hapuarachchi, 2010; Chen et al., 2016; DeZure et al., 2016). Unlike those caused by Zika (ZIKV) and dengue viruses, CHIKV infection is usually associated with high rates of symptomatic infection, including fever, rash, headache, fatigue, myalgia, myocarditis, and recurrent polyarthralgia (Taubitz et al., 2007; Martina et al., 2009; Liu and Zhang, 2016; Rauf et al., 2017). The explosive expansion and severe health burden caused by CHIKV infection have led to a global public health problem.
Previous phylogenetic studies have revealed that CHIKV strains could be grouped into three major geographically distinct lineages, the West African, East/Central/South African (ECSA), and Asian lineages (Volk et al., 2010; Chen et al., 2016). The adaptive mutations that occurred in specific evolutionary lineages have been proven to be associated with transmission and fitness of CHIKV in different hosts and vectors (Tsetsarkin et al., 2007; 2011b). The growing genetic diversity observed in each lineage of CHIKV suggests that the virus may be a major public health threat, presenting a high potential for further reemergence and spreading.
In the current study, we obtained 10 samples of human serum from clinical cases of suspected CHIKV infection in Pakistan and isolated infectious CHIKVs from positive serum specimens using a cell culture procedure. Here, we report for the first time a full-length genome sequence and eight complete envelope (E1) sequences of CHIKVs from Pakistan. Comparison of the E1 sequences suggested that two nonsynonymous substitutions (E99K and Q235H), which occurred at otherwise highly conserved sites, might be associated with a functional effect on the envelope activity. Further phylogenetic analysis indicated that CHIKVs from the Pakistan outbreak originated from CHIKV circulating in the Indian region. This study provides useful information on the CHIKV epidemics in Pakistan, which can help reveal the transmission and evolutionary dynamics of CHIKV in South Asia.
-
Serum specimens were collected from patients in December 2016 to January 2017 during the Karachi CHIKV outbreak in Pakistan. All patients had symptoms of fever, joint pain in limbs, or maculopapular rash and experienced mosquito bites but had no history of traveling abroad. The detailed information on the patients is provided in Table 1.
Serum
numberSampling
locationSampling
datePatient
sexPatient
age (years)Clinical symptoms Traveling
abroad historyMosquito
bite#1 SGHS* 12/23/2016 F 50 Fever, chills, body ache,
headache, joint pain, rashNo Yes #2 SGHS 12/23/2016 F 45 Fever, chills, body ache, headache,
joint pain, anorexiaNo Yes #3 SGHS 12/23/2016 F 50 Fever, chills, body ache, headache,
joint pain, anorexia, rashNo Yes #4 SGHS 12/23/2016 F 18 Fever, chills, body ache, headache,
joint pain, anorexia, rashNo Yes #5 SGHS 12/23/2016 F 40 Fever, chills, body ache, headache,
joint painNo Yes #6 SGHS 12/23/2016 F 65 Fever, chills, body ache, headache,
joint pain, rashNo Yes #7 SGHS 01/05/2017 F 29 Fever, chills, body ache, headache,
joint pain, rashNo Yes #8 SGHS 01/05/2017 F 25 Fever, chills, body ache, headache,
joint pain anorexiaNo Yes #9 SGHS 01/05/2017 M 15 Fever, chills, body ache, headache,
joint pain, anorexiaNo Yes #10 SGHS 01/05/2017 M 14 Fever, chills, body ache, headache,
joint pain, anorexia, rashNo Yes Note: *SGHS: Sindh Government Hospital, Saudabad, Malir, Karachi. Table 1. Information on 10 patients potentially infected with CHIKV in Pakistan
-
Viral RNA (vRNA) was extracted from the serum specimens using a QIAamp viral RNA mini kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions, with a minor modification. Briefly, vRNA was extracted from 45 μL of human serum, and the vRNA extracts were then resuspended in 30 μL of buffer AVE. Total RNA was extracted from infected cells at the indicated time points post-infection using the TRIzol reagent according to the manufacturer’s instruction. All preparations of RNA were stored at –80 °C until use.
-
A real-time reverse transcription–polymerase chain reaction (RT-PCR) assay was performed using a One-Step SYBR PrimeScript RT-PCR kit (TaKaRa, Dalian, China). A SYBR Green-based RT-PCR assay was developed in this study using primers described in our recent study (Liu et al., 2017). All RT-PCR reactions were performed in a total volume of 20 μL, containing 2 μL of an RNA template and 0.4 μL of each CHIKV primer (final concentration, 0.2 μmol/L) in a reaction mixture of the One-Step real-time RT-PCR assay, using a StepOnePlus™ real-time PCR system (Applied Biosystems, Foster City, USA). The RT/amplification conditions consisted of an RT step at 42 °C for 5 min, reverse transcriptase inactivation at 95 °C for 10 s, followed by 40 cycles of PCR at 95 °C for 10 s (denaturation) and 60 °C for 34 s (annealing and extension).
-
Isolation of CHIKV was conducted using C6/36 mosquito cells, similar to the method of ZIKV isolation used in our previous study (Deng et al., 2016a). Briefly, the serum was inoculated directly on C6/36 cells in 35-mm culture dishes. The infected cells were cultured at 28 °C in a 5% CO2 incubator for 1–5 days, depending on the occurrence of cytopathic effects (CPEs) (Figure 1A). Three rounds of serial passages (P1–P3) were conducted for the 10 serum samples.
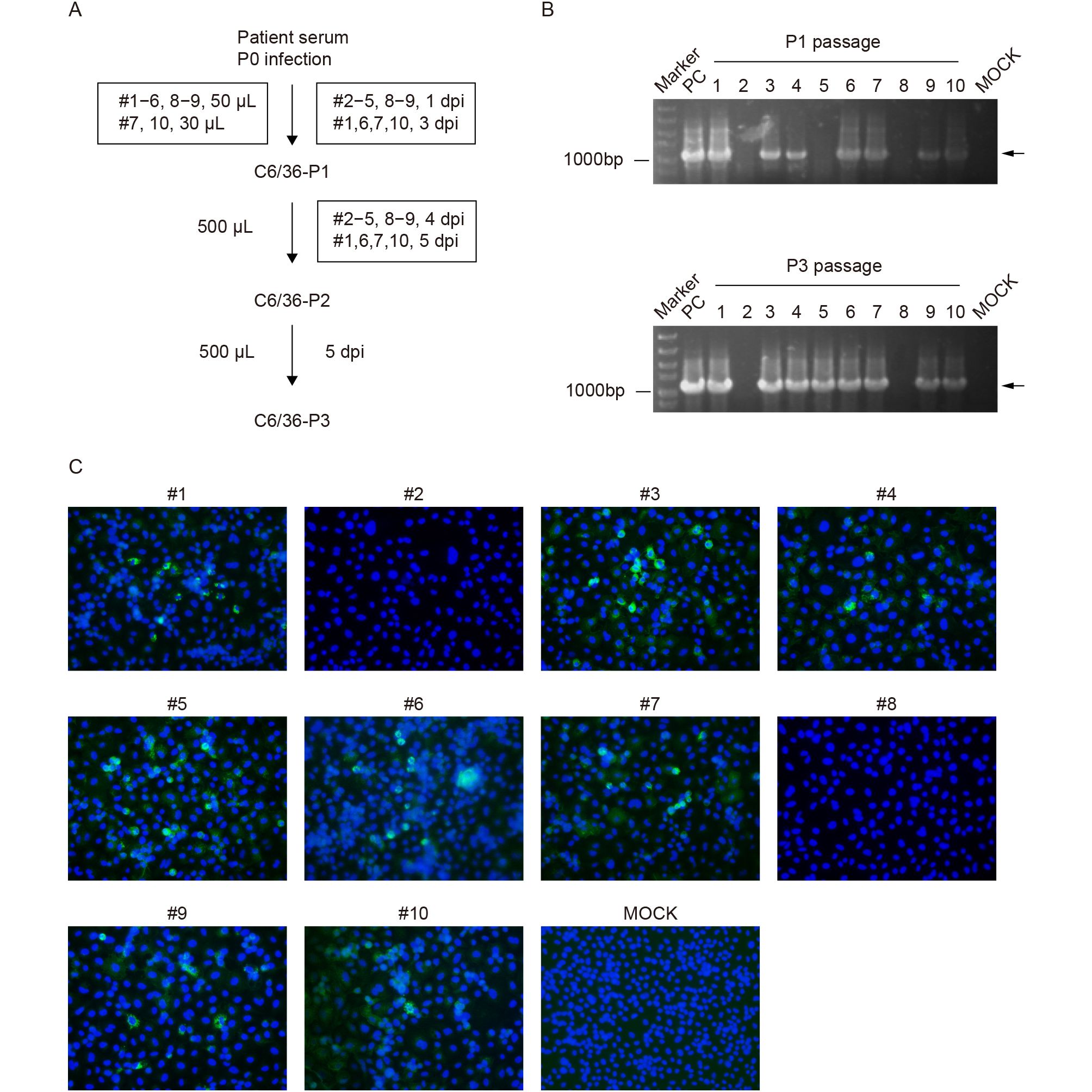
Figure 1. Isolation and identification of new CHIKV strains from Pakistan. (A) Flow chart of virus isolation from 10 human serum specimens on C6/36 cells. (B) Isolation of CHIKVs was confirmed using an RT-PCR assay; the upper and lower panels represent the detection of the first- and third-passage viruses, respectively. (C) Immunofluorescence assay of C6/36 cells, infected with the second passage of the CHIKV strains, at 24 h post-infection.
-
To confirm that CHIKVs were successfully isolated from human serum, a conventional RT-PCR assay and immunofluorescence assay (IFA) were used to detect the presence and replication of CHIKV. The RT-PCR assay was conducted at the first and third passages using the One-Step RT-PCR kit (TaKaRa). Primers specific for the nonstructural protein NSP4–E3 region of the CHIKV genome (forward: 5′-GAAGTGCAGGGTATATCAG-3′; reverse: 5′-GATGCTTGTAGCAGCTGAT-3′) produced an amplicon of about 1,100-bp in length. C6/36 cells infected with the second passage of CHIKV strains were seeded on a chamber slide (Nalge Nunc, Rochester, NY, USA). At 24 h post-infection (hpi), the infected cells were collected, washed with phosphate-buffered saline (PBS), and fixed with 5% acetic acid in acetone. The fixed cells were washed with PBS and incubated with mouse CHIKV E2 polyclonal serum (1:250 dilution with PBS) at 4 °C overnight (Deng et al., 2016b). After washing with PBS, the cells were incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (Proteintech, Wuhan, China), at a 1:125 dilution in PBS, at room temperature for another 1 h. The cells on the slide were mounted with 90% glycerol and examined under an upright fluorescence microscope (Nikon, Tokyo, Japan).
-
Primers targeting the viral peptide 6K gene and 3′-untranslated region (UTR) were used to obtain full E1 sequences of the CHIKV isolates from the human serum samples. The specific sequences of the primers were CHIKV-6K-F: 5′-GAACGAGCAGCAGCCTTTATTT-3′ and CHIKV-3′-UTR-R: 5′-TTCAGCCCTTTGATCTATGCT-3′. A total of 334 E1 sequences, including 333 CHIKV sequences and one Semliki forest virus (SFV, NC_003215) sequence, were obtained from the GenBank database and aligned, using the ClustalX program, with the eight new E1 sequences obtained in our study. The nucleotide and amino acid variations in E1 were estimated using MEGA7. A phylogenetic tree based on the complete genome sequence of CHIKV (Pakistan-03 strain) from this study and 304 CHIKV genome sequences retrieved from GenBank was constructed using the neighbor-joining method in MEGA7. Evaluation of the statistical confidence of each node was based on 1,000 bootstrap replicates.
-
To determine whether the 10 human cases were infected with CHIKV, we first tested the samples by a One-Step SYBR Green-based real-time RT-PCR assay. Using RNA transcripts transcribed in vitro from a full-length infectious clone of CHIKV (Deng et al., 2016b) as a positive control, we found that the mean cycle threshold (Ct) values of the samples ranged from 18.60 (serum #10) to undetermined (serum #8) (Table 2). Based on a standard curve (Table 2), the maximum number of viral genomic RNA copies was estimated to be 2.0 × 107 copies/μL for serum #10. Excluding the undetermined specimen (serum #8), the lowest amount of viral load was detected in serum #2, with the quantity of genomic RNA of 1.2 × 103 copies/μL. Thus, our specific real-time RT-PCR assay revealed that nine serum samples were positive, and these positively diagnosed patients were highly likely to have been infected with CHIKV (Table 2).
Serum specimen number Ct value
(mean ± SD)Mean genomic RNA copy number (copies/μL) #1 19.69 ± 0.0283 1.1 × 107 #2 33.52 ± 0.4243 1.2 × 103 #3 22.40 ± 0.1626 1.8 × 106 #4 21.40 ± 0.0919 3.6 × 106 #5 28.84 ± 0.0141 2.6 × 104 #6 19.14 ± 0.1768 1.6 × 107 #7 21.08 ± 0.1414 4.4 × 106 #8 Undetermined – #9 21.53 ± 0.0849 3.2 × 106 #10 18.60 ± 0.0141 2.0 × 107 Positive control 8.24 ± 0.03606 >109 Negative control Undetermined – Note: The equation of the standard curve constructed for the SYBR Green real-time RT–PCR assay was: Y = –3.496X + 44.289 (R2 = 0.998; efficiency = 93.219%). Table 2. Detection and quantification of CHIKV in 10 human serum specimens from Pakistan
-
A conventional RT-PCR assay was used to confirm the isolation of CHIKV from human serum using total RNA from infected mosquito cells as a template. The targeted DNA bands of approximately 1,100 bp in length were detected for sera #1, #3, #4, #6, #7, #9, and #10 at both P1 and P3 (Figure 1B). The specific band was not detected for the P1 virus from serum #5, while a clearly positive result was obtained for P3 of serum #5 (Figure 1B), indicating that the amounts of vRNA significantly increased from the first to third passages. Together with the number of viral genomic RNA copies (Table 2), the virus load in the original serum specimens played an important role in successful virus isolation. Furthermore, IFA was conducted to verify the replication of CHIKV using an in-house produced mouse polyclonal antibody against CHIKV E2. At the second passage of the serum specimens, IFA-positive cells were observed in all samples, except sera #2 and #8, passaged in C6/36 cells (Figure 1C), which was consistent with the results of the RT-PCR test for P3. Eight new strains of CHIKV, derived from Pakistan, were successfully isolated from sera of infected individuals and were subsequently used for the analysis of variation and molecular evolution of CHIKV.
-
To characterize the genomes of the new CHIKV strains, the E1 gene, whose sequences are among the most frequent in databases and are widely used in phylogenetic analysis, was initially sequenced. Alignment of the E1 sequences (1,320 nucleotides and 439 amino acids) of the eight CHIKV strains showed that there were 1,315 conserved nucleotides and 436 conserved amino acids (i.e., there were three nonsynonymous mutations), accounting for more than 99% of the E1 gene sequence (GenBank accession numbers: MF740874–MF740881). The result suggested that the new CHIKV isolates obtained in our study were closely related to each other.
We further compared CHIKV strains from different evolutionary lineages for the presence of the three nonsynonymous mutations using the entire alignment of 342 E1 sequences, including 341 CHIKV sequences and one SFV sequence used as a reference. The three amino acids in E1 (E99, Q235, and I344), in which nonsynonymous substitutions occurred within the viral genomes of the eight CHIKV strains from Pakistan, were extremely conserved in the E1 sequences of ECSA CHIKV strains. However, the I344V substitution, found in Pakistan-01, was a lineage-specific amino acid substitution of the West African lineage (Figure 2, Table 3). Thus, E99K and Q235H within E1 of CHIKV were not lineage-specific but newly detected substitutions, implying that positive selection might have been driving the evolution of the glycoprotein in CHIKV during the epidemic in Pakistan.

Figure 2. Amino acid sequences and functional domains of membrane protein E1 from CHIKVs and SFV. The domains of the E1 protein were characterized based on the structure of SFV E1 (Lescar et al., 2001; Roussel et al., 2006). The three domains of E1 are shown, with domain I in red, domain II in orange, and domain III in blue. The fusion loop at the tip of domain II is shown in green. The amino acid positions of the ij loop are 219–215 and 232–237 (Roussel et al., 2006). Two nonsynonymous mutations, E99K and Q235H, are marked by red boxes.
Lineage Serum number/Location Strain GenBank ID Nucleotide/amino acid position in E1 295/99 414/138 666/222 705/235 1030/344 ECSA #01/Pakistan Pakistan-01 MF740875 G/E A/Q G/Q G/Q G/V #03/Pakistan Pakistan-03 MF740874 G/E A/Q G/Q G/Q A/I #04/Pakistan Pakistan-04 MF740876 G/E A/Q G/Q G/Q A/I #05/Pakistan Pakistan-05 MF740877 G/E A/Q G/Q C/H A/I #06/Pakistan Pakistan-06 MF740878 G/E G/Q G/Q G/Q A/I #07/Pakistan Pakistan-07 MF740879 A/K A/Q G/Q G/Q A/I #09/Pakistan Pakistan-09 MF740880 G/E A/Q G/Q G/Q A/I #10/Pakistan Pakistan-10 MF740881 G/E A/Q A/Q G/Q A/I –/India 119067 KY057363 G/E A/Q G/Q G/Q A/I Asian –/China CHIKV-JC2012 KC488650 G/E A/Q G/Q G/Q A/I West African –/Senegal 37997 AY726732 G/E A/Q G/Q G/Q G/V Note: The nucleotides and amino acids in bold are variable, including nonsynonymous substitutions, in CHIKV E1 from the ECSA lineage. Strains 119067, CHIKV-JC2012, and 37997 are representatives of the ECSA, Asian, and West African lineages, respectively. Table 3. Variations in the E1 sequences of CHIKV strains from eight human serum samples
-
One CHIKV strain (serum #3) was selected for full genome sequencing (GenBank accession number: MF740874). Subsequently, the sequences of our new CHIKV isolate (named Pakistan-03 strain) and other strains from the National Center of Biotechnology Information database were aligned and used for phylogenetic analysis. A neighbor-joining tree was constructed for the complete genome dataset of CHIKVs. All CHIKV strains were divided into three lineages, ECSA, Asian, and West African lineages (Figure 3). The Pakistan-03 strain was clustered with strain 119067 (KY057363) isolated from a human in India on August 28, 2016. Only 14 variable nucleotides occurred between strains Pakistan-03 and 119067, of which 10 substitutions were synonymous and four were nonsynonymous. This result indicated that CHIKVs spread in Pakistan originated from the circulating CHIKVs of the Indian region, implying that the explosive epidemic of CHIKV in Pakistan could be attributed to the transmission of infested mosquitoes from adjacent areas of India.
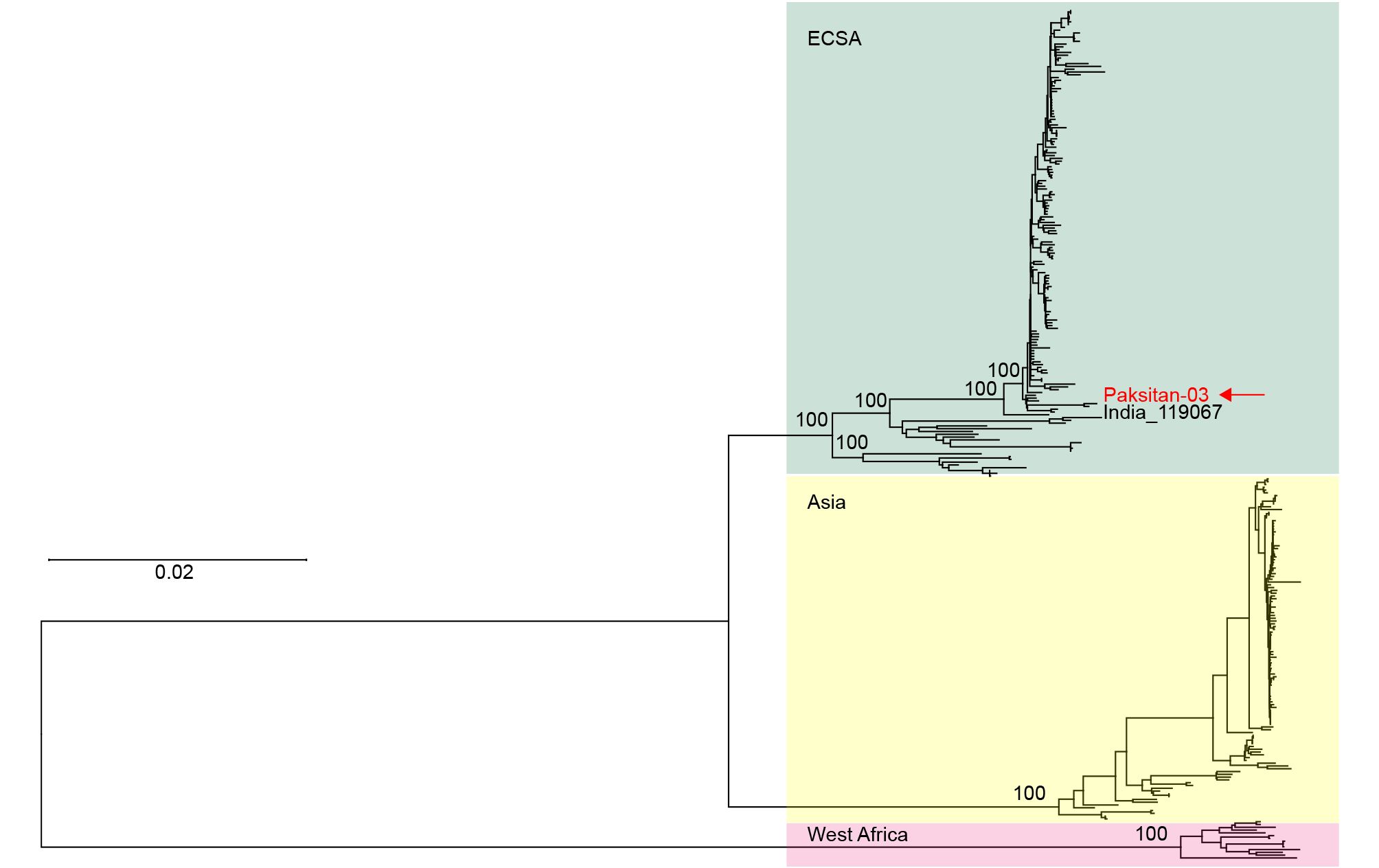
Figure 3. Phylogenetic analysis of CHIKV genome sequences using a neighbor-joining method. The newly isolated and sequenced Pakistan-03 strain is indicated by a red arrow. All CHIKV isolates were divided into three major lineages, East/Central/South African (ECSA), Asian, and West African lineages.
-
The isolation and characterization of CHIKV from mosquito-biting human without travelling history in this study indicate that the viruses are circulating in Pakistan. We established experimental procedures for arboviral detection and isolation in laboratory circumstance and isolated the CHIKV strains from infected residents of Pakistan for the first time.
In isolating the strains of CHIKV, we performed RT-PCR at P1 and P3 and IFA at P2 to confirm CHIKV. The human serum samples probably contained many unknown substances, which resulted in a poor growth and affected the phenotype of the cultured C6/36 cells. Therefore, we only detected CHIKVs at P1 using RT-PCR. In the second round of passaging, the 10 CHIKVs were simultaneously probed using IFA at 24 hpi when CPEs in the 10 cell cultures were not observed yet. Although the percentages of FITC-positive cells were low among infected cells, the IFA results indicated that eight CHIKV strains (#1, #3–7, #9, and #10) were replicating in C6/36 cells, which confirmed replication of the eight CHIKV strains through two rounds of passaging. However, as described in the flow chart of virus isolation (Figure 1A), the CHIKV samples were not passaged simultaneously from P1 to P2. There were two groups (#2–5, #8, and #9 at 4 days post-infection (dpi); #1, #6, #7, and #10 at 5 dpi) because of the different time needed for CPEs to be observed in cells infected with CHIKVs from the two groups. Therefore, we confidently carried out the third round of the passaging experiment for the two groups at different times without RT-PCR testing. The third passage was needed to increase the virus loads of the eight CHIKV strains, which were confirmed by IFA to be positive at P2. Thus, these eight CHIKV strains were simultaneously harvested at 5 dpi and further verified by RT-PCR. The isolation of infectious CHIKVs make it possible to evaluate virus genome replication and the role of specific mutations discussed below.
The newly isolated CHIKV strains are phylogenetically resigned to the ECSA lineage, presenting different replication efficiency compared with a CHIKV strain of Asian lineage (Deng et al., 2016b) previously isolated in our laboratory (data not shown). Two new nonsynonymous mutations (E99 and Q235) were detected within E1 of Pakistani CHIKV. According to the structure of envelope protein E1 of alphaviruses (Figure 2), E99 and Q235 are located in the fusion loop and ij loop of domain II, respectively (Lescar et al., 2001; Roussel et al., 2006). The fusion loop is associated with cell–cell fusion activity, protein folding, and E1 transport out of the endoplasmic reticulum (Levy-Mintz and Kielian, 1991). The spatially adjacent ij loop of E1 has been proven to play a role in the decreased cholesterol dependence of the growth of SFV, which also belongs to alphaviruses (Vashishtha et al., 1998; Chatterjee et al., 2002). Furthermore, an association has been found between the increased fitness of CHIKV in Aedes albopictus and the E1 A226V mutation, which is located in the ij loop at the tip of domain II (Tsetsarkin et al., 2007). Therefore, these nonsynonymous mutations in E1 are likely to be functionally important in regards to the envelope activity. However, it needs to be further investigated whether the differences in E1 sequences affect the virus load and replication.
The recent spread of CHIKV is characterized by reemergence in regions of the Indian Ocean and Asia and migration to Pacific islands and Americas (Chen et al., 2016). Furthermore, adaptive mutations accumulated during circulation have led to distinct evolutionary clades and contributed to rapid distribution and persistent transmission of CHIKV to many countries (Schuffenecker et al., 2006; Tsetsarkin et al., 2007; 2011b). Systematic epidemic surveillance and comprehensive phylogeographic analysis of CHIKV worldwide is important for the prevention and control of the major global health threat. This study sheds a light on the evolution and expansion of CHIKV in South Asia where highly divergent strains are co-circulating, with largely unsampled genetic diversity of CHIKVs from different lineages.
-
This work was supported by International Cooperation on key Technologies of Biosafety along the China–Pakistan Economic Corridor (153B42KYSB20170004), Major State Basic Research Development Program (2013FY113500) and the National Natural Science Foundation of China (81572003 and 31600141). We are grateful to the Core Facility and Technical Support, Wuhan Institute of Virology, Wuhan Key Laboratory on Emerging Infectious Diseases and Biosafety, and Biosafety Level 3 Laboratory, Wuhan Institute of Virology for their helpful supports during the course of the work. We thank the administrator Hao Tang for his hard work in Biosafety Level 3 Laboratory.
-
The authors declare that they have no conflict of interest. The whole study was approved by the ethics committee of the Wuhan Institute of Virology (WIV), Chinese Academy of Sciences (CAS), China. Written consents were obtained from all the patients involved in the study.
-
BZ conceived the experiments. SJ, NJ, HPW, FD, and ALG collected the samples. SQL, XL, YNZ, CLD, and JHL performed the experiments. SQL analyzed the data. BZ and SQL wrote the paper. All authors read and approved the final manuscript.
Detection, isolation, and characterization of chikungunya viruses associated with the Pakistan outbreak of 2016–2017
- Si-Qing Liu 1 ,
- Xiao Li 1,2 ,
- Ya-Nan Zhang 3 ,
- Ai-Li Gao 3 ,
- Cheng-Lin Deng 1 ,
- Jun-Hua Li 1 ,
- Shoukat Jehan 4 ,
- Nadia Jamil 4 ,
- Fei Deng 5 ,
- Hongping Wei 1 ,
-
Bo Zhang
1,,

- Received Date: 04 September 2017
- Accepted Date: 12 December 2017
- Published Date: 21 December 2017
Abstract: The chikungunya virus (CHIKV) is a mosquito-transmitted alphavirus, which has infected millions of people in Africa, Asia, Americas, and Europe since it reemerged in India and Indian Ocean regions in 2005–2006. Starting in the middle of November 2016, CHIKV has been widely spread, and more than 4,000 cases of infections in humans were confirmed in Pakistan. Here, we report the first isolation and characterization of CHIKV from the Pakistan outbreak. Eight CHIKV strains were newly isolated from human serum samples using a cell culture procedure. A full-length genome sequence and eight complete envelope (E1) sequences of CHIKV from Pakistan were obtained in this study. Alignment of the CHIKV E1 sequences revealed that the eight new CHIKV isolates were highly homogeneous, with only two nonsynonymous substitutions found at generally conserved sites (E99 and Q235). Based on the comparison of 342 E1 sequences, the two nonsynonymous mutations were located in well-recognized domains associated with viral functions such as the cell fusion and vector specificity, suggesting their potential functional importance. Phylogenetic analysis indicated that the CHIKV strains from Pakistan originated from CHIKV circulating in the Indian region. This study helps elucidate the epidemics of CHIKV in Pakistan and also provides a foundation for studies of evolution and expansion of CHIKV in South Asia.

 DownLoad:
DownLoad: