












HTML
-
Oncolytic measles virus (OMV) is a potent antitumor agent that preferentially replicates and spreads within tumors as its receptor CD46 (Dörig et al. 1993) is overexpressed on human tumor cells compared with that in their normal nontransformed counterparts (Anderson et al. 2004). Hemagglutinin (H) protein and fusion (F) protein are two kinds of glycoproteins on the envelope of OMV, both of which mediate OMV entry into host cells (Wild et al. 1991). The H protein binds to CD46 and the F protein induces fusion in order to release the nucleic acid into tumor cells (Malvoisin and Wild 1993). The propagation of OMV within tumor cells directly results in cell lysis and indirectly potentiates antitumor immunity (Msaouel et al. 2012). Furthermore, attenuated measles virus (MV) strains have been administered to more than a billion people over the last 60 years, with an excellent safety record (Griffin and Pan 2009). The potent oncolytic effect and safety record of OMV has led to the assessment of OMV-based virotherapy in clinical trials.
It has been reported that more than 90% of Americans have protective titers of anti-measles neutralizing antibodies (NAbs) in their plasma (Lebo et al. 2015), and 85% of children worldwide had received measles vaccine by their second birthday (http://www.who.int), so it is expected that this proportion is similar in other countries. NAbs recognize and bind to the H protein on the envelope of MV (de Swart et al. 2005). As a result, the CD46-binding site of the H protein is blocked; when this occurs in vivo, OMV is cleared by the immune system. Local administration of the virus enables avoidance of the circulating NAbs (Galanis et al. 2010). However, the local delivery route is only applicable to tumors in specific organs or at specific sites.
To overcome this barrier, cells have been used as carriers to deliver OMV to tumor sites. Previous pre-clinical studies have investigated several varieties of candidate cells, including T lymphocytes (Ong et al. 2007), monocytic cells (Peng et al. 2009), mesenchymal stem cells (MSCs) (Mader et al. 2009), endothelial progenitor cells (Wei et al. 2007), and tumor cells (Liu et al. 2010). A phase Ⅰ/Ⅱ clinical trial using MSCs as carriers for OMV is currently ongoing (NCT02068794). Indeed, carrier cells are optimal delivery vehicles, acting as "Trojan horses" (Arap and Pasqualini 2004; Avota et al. 2013), which sophisticatedly prevent immune recognition of the viruses in the circulatory system. Moreover, carrier cells are natural hosts for viruses, and are generally ignored by the immune system (Iankov et al. 2007). In addition to protecting viruses from immune elimination, several types of cells, such as T cells (Ong et al. 2007) and circulating endothelial progenitor cells (Wei et al. 2007), possess tumor-homing abilities. Moreover, carrier cells may serve as sites for viral replication, enabling an increase in the number of viral particles.
The number of viral particles transported to tumor sites by carrier cells may be affected by the loading strategy used, including the initial loading dosage and loading duration. Higher initial loading doses and longer loading durations would be expected to lead to increased viral propagation and enhanced subsequent oncolysis. However, it should be noted that viral antigens expressed on carrier cells are also recognized by host antibodies during the stealth delivery phase (Power and Bell 2007). To date, there have been no systemic studies focusing on the effect of the loading strategy on oncolytic efficacy. In this study, two types of cells, blood outgrowth endothelial cells (BOECs) and the lymphatic cell line Jurkat cells, were employed as carriers to investigate the determinants of viral loading.
-
The human acute T cell leukemia cell line Jurkat (TIB- 152), human non-small-cell lung cancer cell line A549 (CCL-185), and African green monkey kidney cell line Vero (CCL-81) were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). Jurkat cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 2 mmol/L L-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. A549 and Vero cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% FBS, 2 mmol/L L-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin (all from Invitrogen, Carlsbad, CA, USA). BOECs were isolated from adult human peripheral blood, propagated as described previously (Jarmy et al. 2007), and cultured in Endothelial Cell Growth Medium 2 (ECGM-2) supplemented with Endothelial Cell Growth Supplement (C-30160, PromoCell, Heidelberg, Germany) and 10% fetal calf serum (FCS; Biochrom, Berlin, Germany). All cells were maintained in a humidified incubator with 5% CO2 at 37 ℃.
-
Measles virus Edmonston vaccine lineage seed B (MVEdm) and MV-Edm expressing green fluorescent protein (MV-Edm-GFP), kindly provided by S. Russell (Mayo Clinic, Rochester, MN, USA), were propagated in Vero cells. The cells were infected with viruses at a multiplicity of infection (MOI) of 0.02 with Opti-MEM (Gibco, Carlsbad, CA, USA) in a humidified incubator with 5% CO2 at 37 ℃. After 3 h, the medium was replaced with DMEM supplemented with 5% FBS, 2 mmol/L L-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. When 100% of the cell monolayer was fused into syncytia, the cells were scraped off, placed in Opti-MEM, and subjected to three cycles of freezing and thawing to release the viruses. The viral supernatant was centrifuged to remove cell debris and frozen at - 80 ℃. The virus titer was determined by a 50% tissue culture infective dose (TCID50) assay.
-
NAbs were administered in the form of anti-serum. Antiserum was isolated from a healthy donor who had received a measles vaccination. The whole blood was obtained and the serum was separated by centrifugation at 1500 ×g (Heraeus Megafuge 1.0 R, Thermo Scientific, Germany) for 15 min at 20 ℃ and then stored at - 20 ℃. The protocol of this study was approved by the research ethics committee of the Medical School of Nanjing University. The experiments were carried out in accordance with approved guidelines and regulations.
-
Cells were harvested and stained with 0.2% trypan blue (C3601-2; Beyotime Inc., Shanghai, China). Cell numbers and viability were determined using a Countstar Automated Cell Counter (Inno-Alliance Biotech Inc., Wilmington, DE, USA).
-
For the MTT assay, 20 μL of MTT (M5655; Sigma-Aldrich, St. Louis, Missouri, USA; 5 mg/mL) was added intoeach well of a 96-well plate and incubated for 3 h at 37 ℃. Then, the supernatant was removed, 100 μL isopropyl alcohol (12090611516; Nanjing Chemical Reagent Co., Nanjing, China)was added into eachwell, and the plate was agitatedfor 20 min to dissolve the crystals. The absorbance was measured using a Multimode Reader (SMP500-13497-JWYK; Molecular Devices, Sunnyvale, CA, USA) at 570 nm. Cell viability was calculated as the ratio of the absorbance of treated cells to that of the controls (average OD value of treated group/average OD value of control group × 100%).
-
A549 cancer cells were used as the target cells. Cells were seeded at 5 × 103 cells/well in 96-well plates and infected with MV-Edm at MOIs of 0, 0.5, 1, 2, or 4 with or without 10% anti-serum. After 72 h of incubation, the cells were subjected to viability testing using an MTT assay and imaged were acquired by fluorescence microscopy.
-
Next, 104 of each type of carrier cell (Jurkat cells and BOECs) were infected with MV-Edm at the MOI of 2 and, incubated without anti-serum for 4 or 24 h, the cells were harvested for a trypan blue exclusion test. In the absence or presence of NAbs, the cells loaded with MV-Edm were mixed with the target cells (A549 cells) at a ratio of 1:1 (with the same number of viable carrier and A549 cells). After co-incubation for 72 h, the carrier cells in the suspension were removed and the A549 cells were subjected to an MTT assay. A549 cells mixed with uninfected carrier cells were used as controls.
-
Jurkat cells and BOECs were infected with MV-Edm-GFP. After 4, 12, and 24 h, the cells were harvested and washed with PBS. To monitor the expression of GFP, the cells were directly subjected to flow cytometry to analyze the fluorescence intensity (FL1-H). To measure the expression of H protein, cells were incubated with anti-measles H antibody (sc-57913, mouse monoclonal antibody, 1:500 dilution, Santa Cruz Biotechnology, California, USA) for 30 minat4 ℃, and subsequently with mouse IgG1-allophycocyanin (APC) antibody (sc-2888, 1:500 dilution, Santa Cruz) in the dark. After each incubation step, the cells were washed with PBS twice. Finally, the cell pellet was re-suspended in 400 μL PBS and subjected to FACS Calibur (Becton, Dickinson and Company, New Jersey, USA) flow cytometry. The level of H protein expression was indicated by the fluorescence of APC.
-
Student's t-tests were used for statistical analysis. All data are presented as means ± SDs. A value of P < 0.05 was considered to represent statistical significance.
Cells
Viruses
NAbs
Trypan Blue Exclusion Test
3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) Assay
Oncolytic Effect of OMV
Oncolytic Efficacy of OMV-Loaded Carrier Cells
Expression of H Protein and GFP Determined by Flow Cytometry
Statistical Analyses
-
Firstly, we found that MV-Edm induced oncolysis in a dose-dependent manner. However, about 65% of the oncolytic efficacy was abrogated in the presence of antiserum (Fig. 1A). In line with this, MV-Edm-GFP infection and spread was observed in A549 cells in the absence of anti-serum, but completely inhibited by the NAbs (Fig. 1B). These data demonstrate that the anti-serum possesses neutralizing capability against OMV.
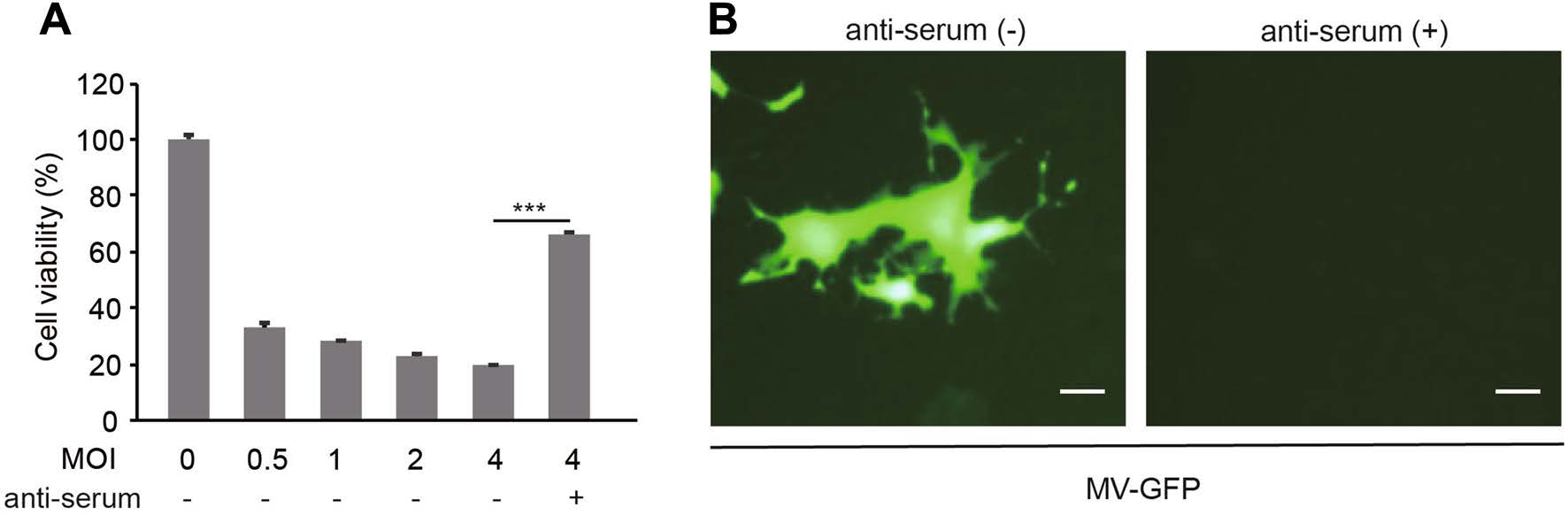
Figure 1. Anti-serum abrogates oncolysis and OMV infection. A A549 cells were infected with MV-Edm at the indicated MOI in the absence or presence of 10% anti-serum. Cell viability was tested by MTT assay at 72 h post-infection. Results are represented as means ± SDs of quadruplicates. Similar results were obtained in three independent experiments. B A549 cells were infected with MV-Edm-GFP at an MOI of 2 in the absence or presence of 10% anti-serum. After 72 h, images were acquired by fluorescence microscopy. Scale bars represent 80 lm. Representative pictures of two independent experiments are shown; ***P < 0.001.
-
Next, we found that the oncolytic capabilities of MV-Edm were sustained in the presence of NAbs when the viruses were preloaded into carrier cells (BOECs and Jurkat cells) (Fig. 2A). Furthermore, the replication and spread of MV-Edm-GFP from carrier cells to adjacent A549 cells were also sustained in the presence of NAbs (Fig. 2B). The results show that carrier cells protect OMV from neutralization by anti-serum.
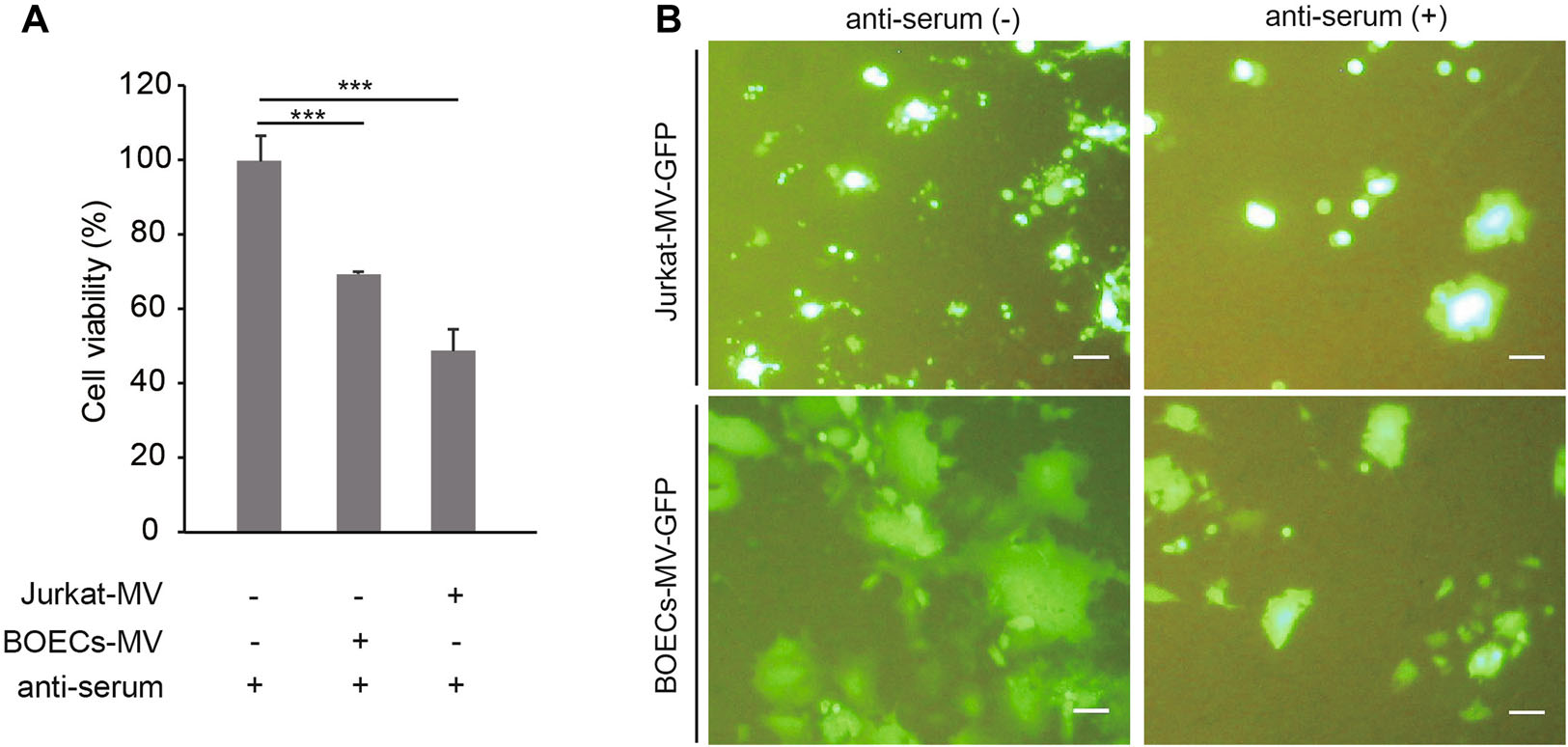
Figure 2. Carrier cells sustain oncolysis and OMV replication and spread in the presence of anti-serum. A BOECs and Jurkat cells were infected with MV-Edm-GFP at an MOI of 2 for 4 h. These carrier cells were then mixed with A549 cells at a ratio of 1:1 and co-cultured for another 72 h in the presence of anti-serum. The viability of the A549 cells was tested by MTT assay. Results are presented as means ± SDs. Similar results were obtained in two independent experiments; ***P < 0.001. B BOECs and Jurkat cells were infected with MV-Edm-GFP at an MOI of 2 for 4 h. These carrier cells were then mixed with A549 cells at a ratio of 1:5 and co-cultured in the presence of anti-serum. After 48 h (BOECs) or 72 h (Jurkat cells), images were acquired by fluorescence microscopy. Scale bars represent 300 lm. Representative images from two independent experiments are shown.
-
Having shown that carrier cells allowed viral replication and oncolysis to occur, we postulated that prolonged loading duration resulted in increased oncolysis by allowing increased time for viral propagation in the carrier cells. Indeed, the oncolytic effect was positively associated with loading duration in the absence of NAbs (Fig. 3A, 3B, filled bars). For instance, when the OMV loading duration in BOECs was prolonged from 4 to 24 h, the oncolytic efficacy was significantly enhanced in the absence of NAbs (Fig. 3B, filled bars). Surprisingly, the enhanced oncolytic effects mediated by prolonged loading duration were completely abrogated in the presence of NAbs (Fig. 3A, 3B, open bars). These results suggest that the viral loading duration influences oncolytic efficacy in the presence of NAbs in vitro.
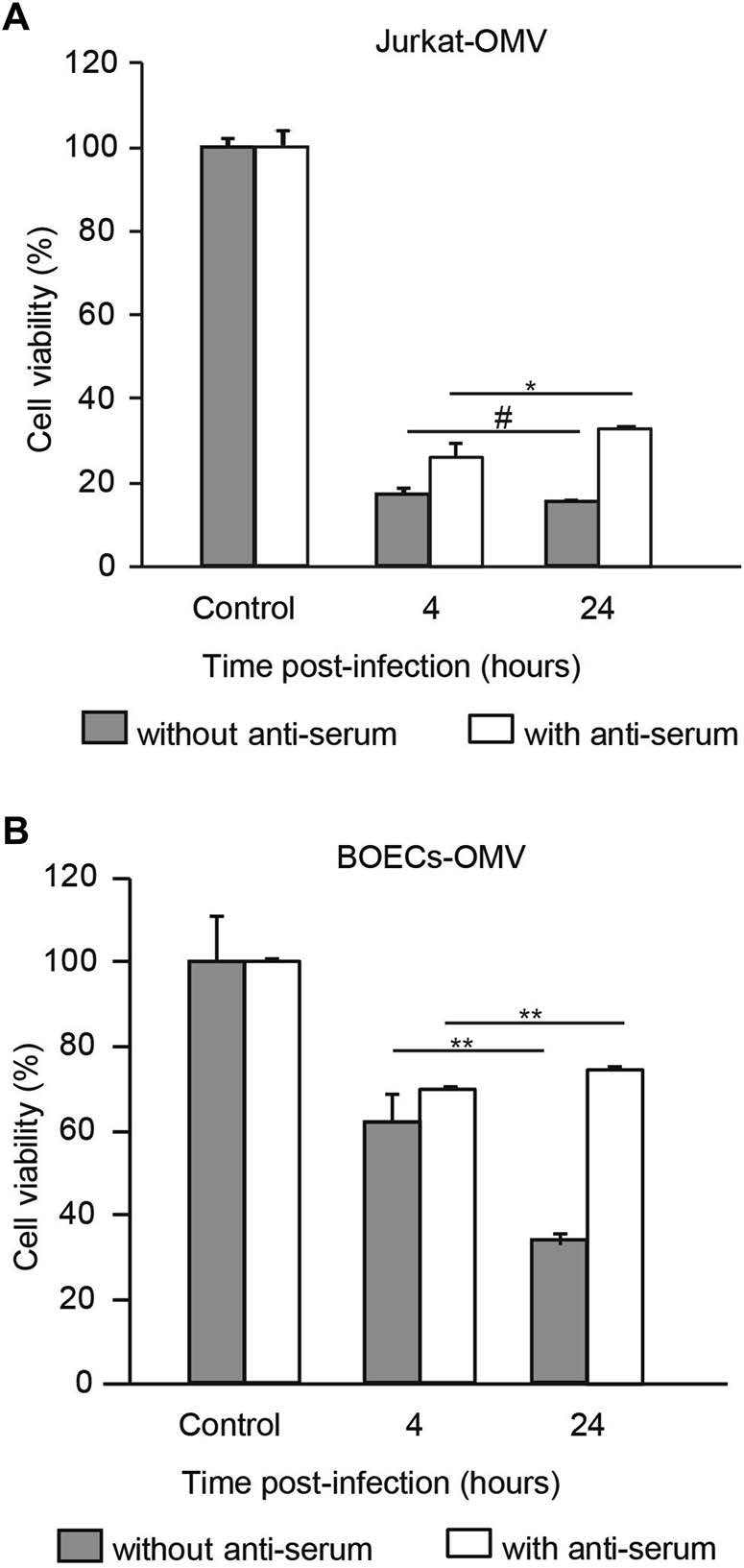
Figure 3. Prolonged loading duration fails to improve oncolytic efficacy in the presence of anti-serum. Jurkat cells (A) and BOECs (B) were infected with MV-Edm-GFP at a MOI of 2 for 4 or 24 h. These carrier cells were then mixed with A549 cells at a ratio of 1:1 and co-cultured for 72 h in the absence or presence of anti-serum. The viability of the A549 cells was tested by MTT assay. A549 cells mixed with uninfected carrier cells were used as controls. Results are presented as means ± SDs. Similar results were obtained in two independent experiments; #P > 0.05, *P < 0.05, **P < 0.01.
-
Next, we sought to investigate the mechanisms by which the loading duration affected the oncolytic efficacy in the presence of NAbs. By counting the GFP-positive cells and measuring the fluorescence intensity of the MV-Edm-GFPloaded carrier cells, we found that viral propagation increased with both increased loading doses and increased loading durations (Fig. 4A, 4B). However, the expression of H protein on both types of carrier cell was mainly determined by loading duration rather than loading dose (Fig. 4C, 4D). For instance, when the loading duration was less than 12 h, increased doses had little impact on the expression of H protein on carrier cells. However, when the loading duration was prolonged to 24 h, H protein expression was dramatically increased on carrier cells, even at a lower loading dose (Fig. 4C, 4D, left panels). These results indicate that a limited viral loading duration is crucial in order to limit the expression of antigenic proteins on carrier cells.
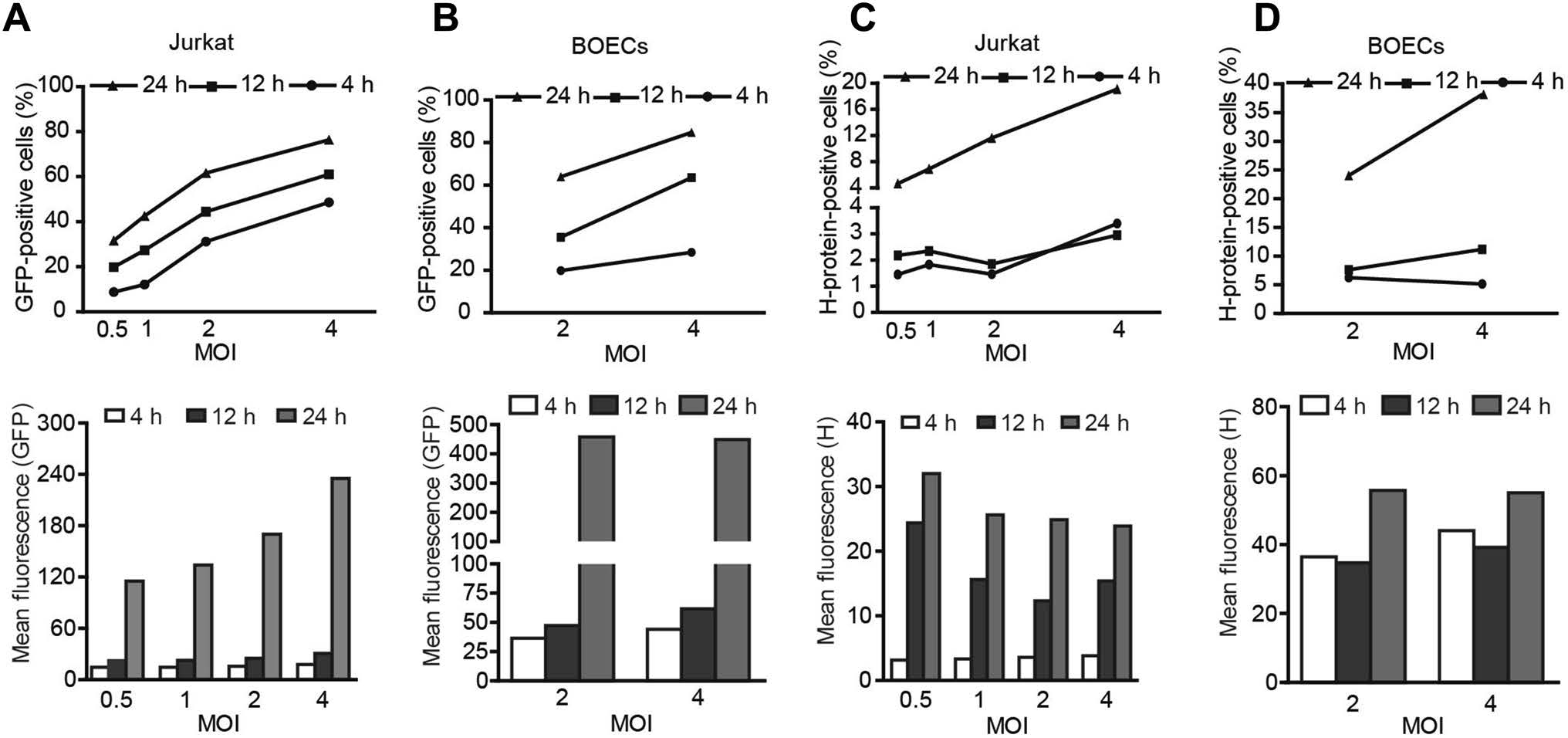
Figure 4. Loading duration predominantly determines the expression of H protein on carrier cells. Jurkat cells and BOECs were infected with MV-Edm-GFP at the indicated MOIs; 4, 12, and 24 h after infection, these carrier cells were subjected to flow cytometry to determine the expression of GFP (A and B) and H protein (C and D). Similar results were obtained in two independent experiments.
-
To further clarify how the NAbs prevented viral transfer from carrier cells to target cells, Jurkat cells were infected with MV-Edm-GFP for 24 h to achieve a high level of H protein expression. Carrier cells were then harvested, incubated with or without NAbs, and mixed with the target cells. In addition, carrier cells were harvested and incubated with target cells for 0.5 or 2 h; then, NAbs were added. We found that syncytial formation was abrogated when the carrier cells were incubated with NAbs prior to mixing with target cells (Fig. 5A, upper panel). However, when the carrier cells were first mixed with target cells for 0.5 or 2 h prior to adding the NAbs, viral transfer was sustained (Fig. 5A, lower panel). In line with this, the mean size of syncytia (which indicates viral transfer) was sustained when NAbs were added after cell-to-cell interactions had occurred (Fig. 5B). These data indicate that the NAbs prevent viral transfer by targeting the H protein prior to the occurrence of cell-to-cell interaction.
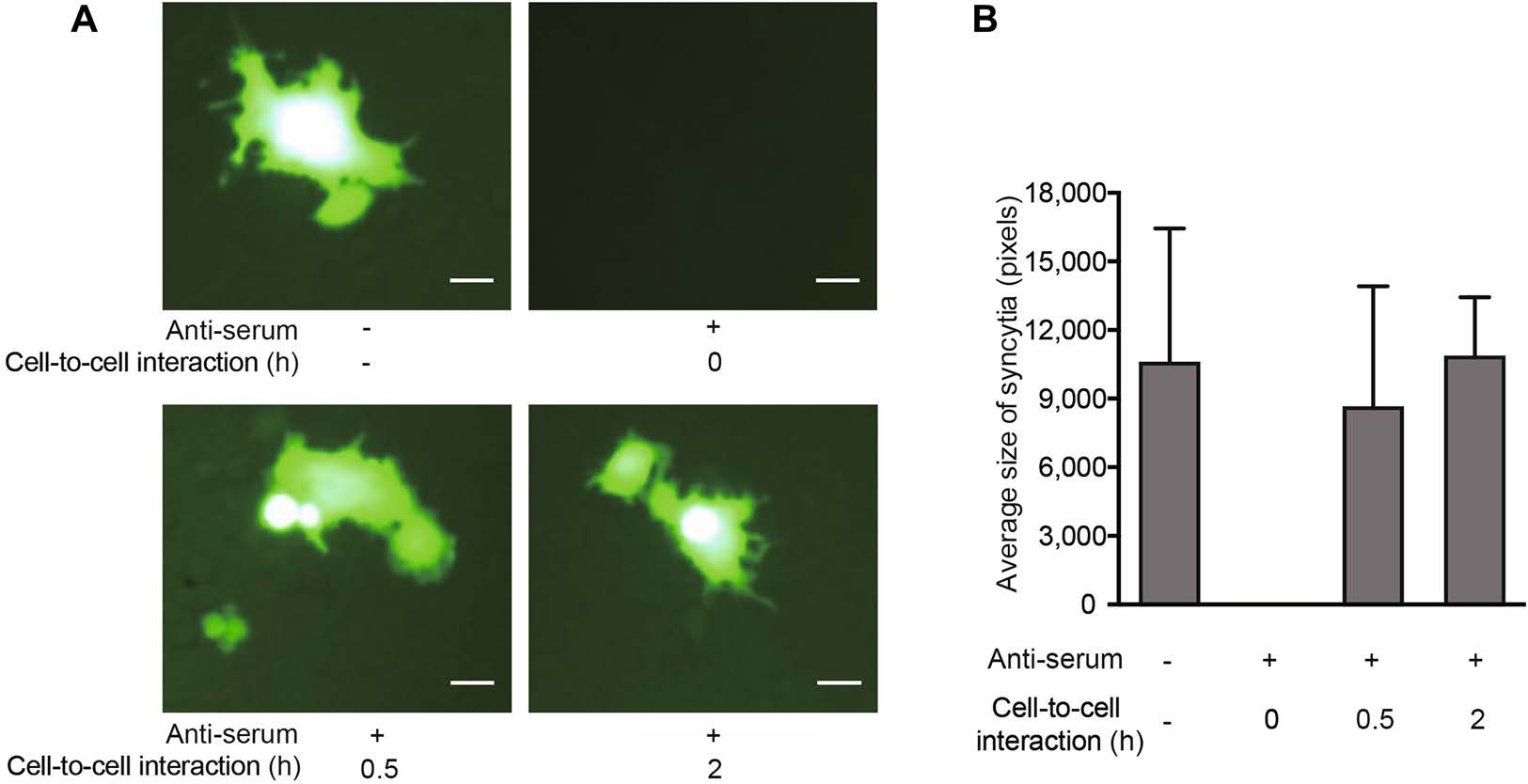
Figure 5. NAbs prevent viral transfer from carrier cells to target cells by targeting H protein prior to cell-to-cell interaction. Jurkat cells were infected with MV-Edm-GFP at an MOI of 0.5 for 24 h. A These carrier cells were then harvested and incubated with or without NAbs prior to mixing them with A549 cells at a ratio of 1:2 (upper panel), or the Jurkat carrier cells were first mixed with A549 cells at a ratio of 1:2 and NAbs were added after 0.5 or 2 h of co-culture (lower panel). Syncytium formation was monitored by fluorescence microscopy 24 h after mixing. Scale bars represent 80 lm. Representative images from two independent experiments are shown. B The syncytium size was quantified using Adobe Photoshop Software and the mean size in each group is shown. Results are presented as means ± SDs. Similar results were obtained in two independent experiments.
Anti-serum Sufficiently Abrogates OMV-Mediated Oncolysis
OMV is Protected by Carrier Cells and is Released from Carrier Cells to Adjacent Tumor Cells in the Presence of Anti-serum
Prolonged Loading Duration Compromises Oncolytic Efficacy of Carrier Cells in the Presence of Anti-serum
H Protein Expression is Mainly Determined by Loading Duration
NAbs Prevent the Viral Transfer from carrier Cells to Target Cells Prior to the Occurrence of Cell-toCell Interaction
-
The oncolytic efficacy of OMV in clinical settings is greatly hampered by pre-existing anti-measles NAbs in patients. Carrier cells have been effectively used for viral delivery (to avoid immune recognition) both in pre-clinical and clinical settings. Therefore, practical loading strategies should be optimized to enhance antitumor efficacy. In this study, we found that the viral loading duration predominantly impacts oncolytic efficacy in the presence of NAbs.
Given that pre-existing NAbs abrogate OMV-mediated oncolysis and the presence of NAbs allows carrier cells to effectively sustain the propagation and spread of OMV (which was confirmed in our previous study (Wei et al. 2007), it was anticipated that a longer loading duration would lead to increased viral yields and, in turn, enhanced oncolysis. Indeed, a prolonged loading duration led to high levels of oncolysis in the absence of NAbs. However, the prolonged loading duration also led to significantly higher expression of H protein on the surface of carrier cells, which were recognized by NAbs, resulting in weakened oncolytic efficacy. Our data show that, at a low MOI, the number of carrier cells expressing H protein increased sharply when the loading duration was increased to 24 h. However, with a given loading duration (e.g. 12 h), there was no obvious change in the H protein level at either low or high MOI. Thus, the expression of H protein by loaded carrier cells was predominantly determined by the loading duration rather than by the loading dose.
It has been shown that NAbs recognize and bind to the H protein on the envelope of MV (de Swart et al. 2005). Therefore, the loading duration is a critical factor when considering strategies aimed at shielding carrier cells from NAbs. During the early stage after virus loading, carrier cells do not display H protein on their surfaces; therefore, these cells are able to associate with target cells even in the presence of anti-measles NAbs and subsequently express the H protein in a fusion synapse (which is protected from NAbs). Conversely, when H protein is already expressed at high levels on the carrier cells prior to mixing them with the target cells, NAbs are able to block the interaction. This is supported by our findings that NAbs efficiently abrogate viral transfer from carrier cells that express high levels of H protein, but fail to block viral transfer when cell-to-cell interactions have already occurred between the loaded carrier cells and the target cells. These results indicate that NAbs prevent viral transfer by targeting H protein prior to the occurrence of cell-to-cell interaction.
In this study, we also observed the differential cell killing effects between carrier cells such as BOECs and Jurkat cells, even under the same loading conditions. For instance, MV-Edm-loaded Jurkat cells exerted stronger oncolytic effects than MV-Edm-loaded BOECs. This difference may be attributable to the carrier cell-specific rates of viral propagation, release, and spread. Therefore, to achieve more effective antitumor effects, appropriate carrier cells should be selected for a given oncolytic virus.
The use of carrier cells provides a practical solution that enables oncolytic viruses to circumvent host anti-viral immunity. It should be noted that carrier cells may be recognized by the NAbs if viral antigens are presented on the cell membrane to a sufficient degree, transforming the cells into "exposed Trojan horses". Our work demonstrates that the loading duration is the predominant determinant of H protein expression in carrier cells. With respect to each unique Trojan horse/oncolytic virus combination, the optimal loading strategy should be systematically investigated to achieve the maximal oncolytic effect for clinical applications. The present findings represent an important step toward that milestone.
-
The authors thank Prof. Dr. Stephen J. Russell for providing modified oncolytic MV strains. This study was supported in part by the National Natural Science Foundation of China (81472820, 81773255, 81071860 and 81602702), Jiangsu Special Program for Clinical Medical Science and Technology (BL2014054), the Natural Science Foundation of Jiangsu Province of China (BK20160126), and the Fundamental Research Funds for the Central Universities (021414380223 and 14380336/1-2). Six talent peaks project in Jiangsu Province to JW.
-
JW designed the study; CX, MX, GM and CL performed the experiments; CX and MX analyzed the data. CX and JW wrote the manuscript. JW and AJ finalized the manuscript.
Acknowledgements
Author Contributions
-
The authors declare that they have no conflict of interest.
-
The protocol of this study was approved by the Research Ethics Committee of the Medical School of Nanjing University. The experiments were carried out in accordance with approved guidelines and regulations. All participants were provided with written informed consent.

 DownLoad:
DownLoad: