














HTML
-
Myristoylation is the covalent attachment of myristic acid, a 14-carbon saturated fatty acid, to the N-terminal glycine of proteins expressed in eukaryotic cells (Boutin 1997; Farazi et al. 2001; Gordon et al. 1991). Although often referred to as post-translational modification (PTM), myristoylation usually occurs co-translationally (Wilcox et al. 1987). This process is catalyzed by enzymes prevalent in eukaryotes, including N-myristoyl transferase (NMT) (Udenwobele et al. 2017). The binding site of myristic acid includes a consensus sequence that requires a serine (Ser) or threonine (Thr) residue in the fifth position, in addition to a Gly residue at the N-terminus (G1X2X3X4S/T5X6X7X8) that facilitates myristoylation of many proteins (Johnson et al. 1994).
A large number of proteins with diverse functions are modified by N-myristoylation (Thinon et al. 2014). The prevalence of N-terminally myristoylated proteins in eukaryotes has been estimated to be between 0.5% and 3% of the cellular proteome, depending on species and the predictive model used (Martinez et al. 2008; Maurer-Stroh et al. 2002). These proteins have a broad range of functions and include protein kinases and phosphatases, Gα proteins, nitric oxide synthase (Braam and Verhaar 2007), ADP-ribosylation factors (ARFs), calcium-binding proteins and membrane or cytoskeleton-associated structural proteins such as MARCKS (Raju et al. 1995; Resh 1999). Hence, myristoylation plays an essential role in multiple cellular mechanisms.
In addition to cellular functions, myristoylated proteins are involved in a wide variety of physiological activities in viruses, including replication, assembly, and infection. For example, covalent linkage of myristic acid to an N-terminal glycine residue in Pr55gag, the precursor of major structural proteins in human immunodeficiency virus 1 (HIV-1), facilitates an essential step in virus assembly and propagation (Bryant and Ratner 1990). Myristoylation is also critical for the function of the Nef protein in HIV (Geyer and Peterlin 2001; Giese et al. 2006; Zhu et al. 2017). These studies indicate that during HIV infection, the virus anchors myristoylated Nef to the plasma membrane, where it binds to and degrades surface receptors (Hill and Skowronski 2005; Mariani and Skowronski 1993; Peng and Robert-Guroff 2001; Udenwobele et al. 2017). Myristoylation also occurs in murine leukemia virus (MLV) (Rein et al. 1986), Mason-Fisher virus (MPMV) (Rhee and Hunter 1987), and Rouse's sarcoma virus (Wills et al. 1989), suggesting that for any of these viruses myristoylation is a necessary step for viral infection. Moreover, the non-myristoylated spleen necrosis virus exhibits inhibition of virus replication, aberrant proteolytic processing, interruption of virion assembly, and decreased releasing of virus from cells (Weaver and Panganiban 1990). Treatment of Lassa virus-infected cells with various myristoylation inhibitors also drastically reduces efficient Lassa virus replication (Strecker et al. 2006), further indicating that this protein modification is crucial for viral functioning.
Myristoylation also plays a crucial role in picornaviruses. Host ribosomes translate the RNA genome of picornaviruses into a single polyprotein, precursor protein P1 (Jiang et al. 2014). The N-terminal glycine (position 2) of the newly formed P1 becomes rapidly modified by covalent linkage to myristic acid (Chow and Moscufo 1995; Chow et al. 1987; Paul et al. 1987). Capsid assembly begins with cleavage of P1 into VP0, VP3, and VP1 by viral protease, and assembly into protomers, which then assemble into pentamers and subsequently into an icosahedral capsid that encloses the viral RNA genome; finally, VP0, in most picornaviruses, is processed into VP4 and VP2, after which mature infectious virions are released (Mousnier et al. 2018). The myristic acid moiety may potentially function in the early and late processes of particle assembly (Marc et al. 1990; Moscufo et al. 1991), with two possible functions for myristoylation having been proposed during early particle assembly. First, the myristoylation of VP0 interacts with the membrane replication complex (Martin-Belmonte et al. 2000). Second, the myristic acid component promotes the protomer interaction of the pentameric assembly (Ansardi et al. 1992; Lee and Chow 1992), similar to functions proposed for myristoylation during the pentamer formation of foot-and-mouth disease virus capsid (Goodwin et al. 2009). VP0 myristoylation also appears to be important in the later period of poliovirus (PV) assembly during virus maturation (Ansardi et al. 1992; Marc et al. 1990; Moscufo et al. 1991).
Enterovirus 71 (EV71) is a member of Picornaviridae family and is an important pathogen in hand, foot, and mouth disease. Further, neurological complications may develop with the potential to inflict permanent morbidity or even fatality, thereby posing significant public health concerns, specifically for infants and children (Alexander et al. 1994; Chumakov et al. 1979; Lum et al. 1998). The EV71 VP4 is also co-translationally linked to myristic acid at its amino-terminal glycine residue (position 2) (Chow et al. 1987); however, the role of myristoylation in EV71 remains largely unknown.
In this study, we constructed myristoylation-deficient EV71 models with Gly-to-Ala mutation (G2A), and analyzed viral genome RNA replication and assembly in both the reporter pseudovirus system and live virus system. Using these virus models, we found that loss of myristoylation had a severe effect on viral infectivity. Furthermore, we examined the interaction between myristyl groups and membrane structure, as well as the possible mechanism of myristoylation in the EV71 virus.
-
The SV40-transfected human embryonic kidney 293 cell line HEK293T was cultured as a monolayer in Dulbecco's Modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS). HEK293S cells stably expressing SCARB2 (Li et al. 2013), which is the EV71 receptor, were cultured as monolayers in DMEM supplemented with 10% FCS and 1.25% puromycin as selective media for SCARB2 expression.
-
We employed the previously published plasmid, GFP-P1-WT (Jin et al. 2012), composed of green fluorescent protein (GFP) and EV71 P1 genes with a linker coding the EV71 2Apro cleavage motif (KGLTTY↓G, where the arrow indicates the cleavage site). To abolish the myristoylation signal, we generated a G2A mutation by sitedirected mutagenesis of the codon specifying the second amino acid of P1, a glycine (which is also the second amino acid of VP4) (Krausslich et al. 1990). P1 of the EV71 introduced a glycine to alanine (G2A) mutation using primers 5'-CTAAACATGGCTTCGCAAGTG-3' and 5'-CACTTGCGAAGCCATGTTTAG-3', and the mutant sequence was confirmed by sequence analysis by Comate Bioscience Company Limited. The plasmids, pcDNA3.1-T7 RNA Pol and pBS-T7-EV71-luciferase, were constructed as described previously (Jin et al. 2013). T7 RNA polymerase expression plasmid (pT7 RNA Pol), was used for expressing T7 RNA polymerase. The pT7-EV71-luciferase replicon plasmid is a reversely transcribed fulllength EV71 genome encoding the firefly luciferase gene in place of the P1 gene, which can be packaged in the nuclei of virus particles; and EV71 capsid expression plasmid (pGFP-P1-WT or pGFP-2A-P1-G2A), which expresses the EV71 capsid proteins as a fusion protein with GFP under the control of the cytomegalovirus (CMV) promoter, and can trans-encapsidate the full-length EV71 genome (Jin et al. 2012).
EV71 reporter pseudoviruses were produced following cotransfection of 293T cells with three plasmids pGFP-P1-WT or pGFP-2A-P1-G2A, pT7-RNA-pol and pT7-EV71-luciferase (Arita et al. 2006; Jin et al. 2013; Xu et al. 2014). Briefly, in a 6 wells plate, a 70% confluent monolayer of 293T cells was co-transfected with 1 μg of each of pT7-RNA-pol, pT7-EV71-luciferase and pGFP-2A-P1 or pGFP-2A-P1-G2A in 10 μL of Lipofectamine2000 reagent (Invitrogen, California, USA) and incubated at 37 ℃ in 1 mL DMEM (Jin et al. 2012). The cells were washed, and 10% FCS-DMEM was added to the cells at 6 h posttransfection and then incubated for 48 h. The supernatant (containing the virions) from cell cultures was then harvested and stored at - 80 ℃.
-
The plasmid pwsk-T7-EV71 (WT) containing full-length EV71 genome (C4b) was constructed as described previously (Cao et al. 2019). To generate the G2A mutation, a similar protocol as that described for G2A reporter luciferase pseudoviruses was followed. This was facilitated by cloning the 5'-terminal sequences of EV71 into a vector suitable for mutagenesis. The SphI-BamHI DNA fragment (nucleotides [nt] 1751–2667) of pwsk-T7-EV71 was inserted into the polylinker region of plasmid VR1012 (Invitrogen, USA). The primers used for the mutation were the same as those used to mutate the G2A reporter luciferase pseudoviruses at the second amino acid of P1 (glycine to alanine, G2A). The mutant sequence was confirmed by sequence analysis. The mutant SphI-BamHI fragment of VR1012 (corresponding to nt 1751–2667 of the pwsk-T7-EV71) was cloned back into the BamHI-SphI fragment of plasmid pwsk-T7-EV71, and the mutations were verified by sequence analysis.
EV71 live viruses were produced through cotransfecting 293T cells with pT7-EV71-WT or pT7-EV71-G2A, and pT7-RNA-pol (Cao et al. 2019). Briefly, in a 6 wells plate, a 70% confluent monolayer of 293T cells was co-transfected with 1 μg pT7-RNA-pol and pT7-EV71-WT or pT7-EV71-G2A in 6 μL of lipofectamine2000 reagent (Invitrogen) and incubated at 37 ℃ in 1 mL of DMEM. The cells were washed, and 10% FCS-DMEM was added to the cells at 6 h post-transfection and incubated for 48 h. The supernatant of cells was then harvested and stored at - 80 ℃.
-
To observe the intracellular distribution of proteins with or without myristoylation, we constructed the plasmid VP4-WT-GFP and VP4-G2A-GFP, which was composed of VP4-WT or VP4-G2A genes and GFP. Briefly, the VP4-G2A gene was synthesized by PCR with the template for the pGFP-2A-P1-G2A plasmid, and the template for VP4-WT genes was pGFP-2A-P1. At the 5' end of the VP4 sequence, we created the NotI restriction sites by PCR, while at the 3' end we synthesized 20 nucleotides, which were the same as the 5' end of GFP gene. The GFP gene was also synthesized by PCR with the template of the pGFP plasmid. At the 3' end of the GFP sequence, we created the XbaI restriction sites by PCR. Vector VR1012 cleaved at the NotI, and XbaI site and three fragments: VP4-WT or VP4-G2A, GFP, and NotI-XbaI fragment were isolated in a low gelling temperature agarose gel. The VP4-WT or VP4-G2A and GFP fragments were mixed for homologous recombination as instructed with In-Fusion® HD Cloning Kit (Takara) user manufacture. Then the VP4-WT-GFP or VP4-G2A-GFP fragment was inserted into the NotI-XbaI fragment of plasmid VR1012 and plasmid pVP4-WT-GFP or pVP4-G2A-GFP was verified by sequence analysis.
-
The WT-VP4 or G2A-VP4 full-length peptides, the WTVP4 or G2A-VP4 short peptides including the first 20 amino acids of VP4 (EV71-WT or G2A-VP4-20aa), and HRV VP4 confirmed to induce permeability in model membranes (Zauner et al. 1995) were obtained from GL Biochem (Shanghai, China). The sequences of the polypeptides are shown in Table S1.
The purity of the peptides was determined by analytical reverse-phase high-pressure liquid chromatography (RPHPLC) using a C18 column, and peptide masses were confirmed by time-of-flight mass spectrometry performed with an LTQ Orbitrap Velos mass spectrometer (Thermo, CA, USA). Purified peptides were stored at - 80 ℃ (Zauner et al. 1995). The data is presented in Supplementary Fig. S1.
-
Whole-cell extracts were prepared by cell lysis in RIPA buffer for 30 min on ice and clarified at 20, 000 ×g for 5 min at 4 ℃. The supernatants were added to SDS-PAGE sample buffer, boiled, and separated in 13.5% polyacrylamide gels. Proteins were electro-transferred onto PVDF membranes for Western blot analysis. Membranes were probed with rabbit anti-VP3, anti-VP2, and anti-VP1 polyclonal antibodies (1:1000, GeneTex, USA) in PBS with 1% skim milk, followed by probing with a corresponding alkaline phosphatase (AP)-conjugated secondary antibody (Sigma, St. Louis, MO, USA) that was diluted 1:10, 000. And the immunoreactions were visualized with nitro blue tetrazolium (NBT, Dinguo, Beijing, China) and 5-bromo-4-chloro-3indolyl phosphate (BCIP, Dinguo) solutions.
-
To perform transmission electron microscopy (TEM), WT or G2A reporter pseudoviruses were obtained from transfected 293T cells. Briefly, after two rounds of freezing and thawing, the transfected 293T cell lysates were filtered through a 0.22-μm membrane. WT or G2A reporter pseudoviruses were purified from the resulting suspension using DEAE-Sepharose (Amersham Pharmacia Biotech, Piscataway, NJ, USA) (Horodniceanu et al. 1979), followed by centrifugation at 100, 000 ×g for 2.5 h at 4 ℃ in a Beckman SW40 rotor through a 1 mL 30% sucrose cushion. The pellet was washed thrice with distilled water and dissolved in 500 μL phosphate-buffered saline (PBS). Samples were examined by TEM (JEM-1220; JEOL Datum, Tokyo, Japan) at an acceleration voltage of 80 kV, and the images were obtained at 200, 0009 magnification (Jin et al. 2013).
-
HEK293T cells were co-transfected with pT7 RNA Pol (Xu et al. 2014) and pT7-EV71-WT or G2A using lipofectamine 2000 follow the instructions provided by the manufacturer (Invitrogen, Carlsbad, CA, USA). After 48 h, the cells were collected with 1 × PBS and centrifuged at 300 ×g for 5 min, after which the supernatant was discarded. Next, 1 mL acetaldehyde fixing solution was added to precipitate the cells into a spherical shape. The samples were sent to the China National Biological Products Institute, and the high-pressure frozen tablets were then observed by transmission electron microscopy.
-
Reporter pseudoviruses were collected from the supernatants, which were serially diluted fivefold in DMEM (10% FCS) and used to inoculate 293S cells (4 × 104 cells per well) in quadruplicate in 96-well plates (BD Biosciences). These cultures were incubated for 18 h at 37 ℃, after which the cells were harvested for luciferase activity measurement using a luciferase assay kit (Promega, USA) according to the manufacturer's recommendations. Briefly, cells were lysed in 100 μL reporter lysis buffer (Promega, USA), and luciferase activity in cell lysates (100 μL per well) was measured using a VICTORTM × 2 Multilabel Plate Reader (Perkin Elmer, UK).
-
293S cells were added to the plates at 90% confluency. A virus with the equal amount of viral RNA load was used to inoculate individual wells of the 6-well plates. The plates were incubated at 37 ℃ with 5% CO2 for 1 h, with agitation every 15 min. The virus inoculums were then discarded; cells were washed thrice with 1 × PBS and inoculated with 2 mL DMEM medium containing 10% FCS. The cells were observed daily.
-
In a 12-well plate, a 70% confluent monolayer of 293S cells was infected with live-WT and G2A viruses with an equal amount of RNA. At 1 h post-infection, the virus supernatant was discarded, fresh medium was added and incubated for 24 h. These cells were denoted as the P0 generation cells (P0) and were resuspended, added to a fresh 6-well plate with fresh medium added to allow for cell proliferation. After 24 h, the cells covering the 6-well plates were denoted the P1 generation cells (P1), which were then placed in a fresh T25 flask with fresh medium to continue the culture. After 24 h at 37 ℃, the cells that covered with the T25 flask were designated the P2 generation (P2). Half of the P0 and P1 cells were used to measure the amount of viral RNA, while the other half was used for cell passaging. Half of the P2 cells were used for measuring viral RNA and the other half were added to a new well in a 6-well plate and used for immunofluorescence assaying once the cells had attached.
-
Live WT viruses with equal amount viral RNA infected 293S cells treated with a final concentration of 100 μmol/L 2-HMA or without 2-HMA. After cells without 2-HMA produced CPE, Progeny non-myristoylated viruses were then collected from cells with 2-HMA, and used in infection for construction of a growth curve. WT, G2A live virus and Progeny non-myristoylated viruses containing an equal amount of viral RNA infected 293S cells. NH4Cl at a final concentration of 40 mmol/L was used to treat 293S cells 2 h before infection with live WT virus, and after infection. Negative viral RNA content was then measured.
-
The total RNA of cells infected with WT or G2A virus or reporter pseudovirus was extracted using the RNAprep pure CellorBacteria kit (TIANGEN Biotech Co., Ltd, Beijing, China) according to manufacturer's instructions. cDNA was synthesized using the Prime ScriptTM RT reagent kit with gDNA Eraser (Takara, Japan), and cDNA levels were quantified using real-time PCR (SYBR GreenTM Premix Ex TaqTM, Takara, Japan) using primers EV71-5'UTR-F (5'-TCCTCCGGCCCCTGA-3') and EV71-5'UTR-R (5'-AATTGTCACCATAAGCAGCCA-3') targeting the EV71 5' UTR. The GAPDH gene was used as an internal control to normalize the total RNA in the samples.
-
293S cells infected with WT and G2A live virus were fixed with 4% paraformaldehyde for 15 min and treated with 0.2% Triton X-100 for 15 min, blocked with 1% bovine serum albumin for 1 h, and subsequently incubated with an anti-EV71 VP3 rabbit polyclonal antibody prepared as previously reported (Zhang et al. 2012) for 2 h, followed by reaction with a FITC-goat anti-rabbit antibody (Bioss Biotech Co., Ltd, Beijing, China) for 1 h. The cell nuclei were stained with DAPI (Key GEN Biotech Co., Ltd, Nanjing, China). Fluorescence was recorded using the Zeiss LSM 710 (Zeiss, Jena, Germany).
-
The release of viral mRNA into the cytoplasm is a prerequisite for enabling replication of viral RNA to allow for replication of the viral negative-strand RNAs. Hence, the amount of negative-strand RNA is reflective of whether viral mRNA is released into the cells (Zhang et al. 2017). Quantification of negative-strand genomic RNA replication was performed as previously described (Zhang et al. 2017), and the total RNAs of the infected cell samples were extracted and subjected to RT-PCR quantification as described previously (2.10). In brief, cDNA was synthesized with a Prime ScriptTM RT reagent kit with gDNA Eraser (Takara, Japan) using a primer specific to the negative-strand genomic RNA (1F: 5'-TTAAAACAGCCTGTGGGTTG-3') (Zhang et al. 2017). The cDNA synthesized from negative-strand genomic RNA was then quantified with real-time PCR using 50 UTR specific primers. The GAPDH gene was used as an internal control to normalize the total RNA of the sample.
-
293T cells were grown until subconfluency in 24-well plates was achieved. Cells were transfected with pVP4-WT-GFP or pVP4-G2A-GFP and incubated for 24 h, after which the cells were washed with PBS, fixed with 4% paraformaldehyde, and permeabilized with PBS containing 0.5% saponin. Samples were blocked for 15 min in 0.5% fish gelatin in PBS. Cell nuclei were counterstained with DAPI. All steps were carried out at room temperature. The samples were examined with an LSM 700 confocal laser scanning microscope (Zeiss), and images were processed with ImageJ.
-
Cytoplasmic and membrane fractionations were examined as previously described (Colomer-Lluch and Serra-Moreno 2017). Briefly, 5 × 106 293T cells were transfected with 5 μg pVP4-WT-GFP or pVP4-G2A-GFP plasmid for 24 h. Cells were then lysed, and sequential extraction of proteins from specific cellular compartments and/or organelles was performed using the ProteoExtract® Subcellular Proteome Extraction kit (S-PEK, Millipore, Bedford, MA USA) following the manufacturer's instructions. Cytoplasmic fraction (F1) and membrane protein fraction (F2) were analyzed via western blotting using an anti-GFP monoclonal antibody. Whole-cell extracts were confirmed by blotting for total protein. The membrane and cytoplasmic fractions were determined by detecting GAPDH (cytoplasmic marker).
-
The ability of the compounds to disrupt model lipid membranes was assayed by the release of a fluorescent dye from liposomes loaded with a self-quenching concentration of carboxyfluorescein (Wagner et al. 1992). To verify the membrane interaction of myristoylation, a liposome leakage assay was performed as described previously (Zauner et al. 1995). Briefly, liposomes were prepared from egg phosphatidylcholine (Avanti Polar Lipids) by reversephase evaporation with an aqueous phase of 100 mmol/L carboxyfluorescein (CF)-50 mmol/L NaCl and extruded through a 100-nm polycarbonate filter to obtain a uniform size distribution. The liposomes were separated from unincorporated material by gel filtration through Sephadex G-25 with an iso-osmotic buffer (200 mmol/L NaCl, 25 mmol/L HEPES pH 7.3). Next, 80 μL of liposome solution was added to 80 μL peptide for a final concentration of 30 nmol/L in water in a 96-well microtiter plate (final lipid concentration, 25 μmol/L) and assayed for CF (Carboxyfluorescein) fluorescence at 485–520 nm on a VICTORTM × 2 Multilabel Plate Reader (Perkin Elmer, UK). The test time was 30 min, during which the fluorescence value was recorded every minute.
-
All experiments were repeated thrice independently. The values were expressed as the mean ± standard deviation (SD). Statistical analysis was performed using a one-way ANOVA to compare the differences in test values between the experimental and control groups. Statistical significance represented by asterisks was marked correspondingly in the figures, where *P < 0.05, **P < 0.01, ***P < 0.001, ns: non-significant.
Cells
Plasmid Construction and Virus Production via Reverse Genetics Approach
Production of Wild Type (WT) or G2A Reporter Pseudoviruses
Production of WT or G2A Live Virus
VP4-WT-GFP or VP4-G2A-GFP Expression Plasmid
Peptide Synthesis
Western Blotting
Transmission Electron Microscopy
High-Pressure Cryo-electron Microscope
Luciferase Reporter Gene Assay
Cytopathic Effect Assay (CPE)
Infected Cells Blind Passage
Viruses Infected Cells with Inhibitor
Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
Immunofluorescence Assay
Quantification of Negative-Strand Genomic RNA
Confocal Microscopy
Cytoplasmic and Membrane Fractionations
Liposome Leakage Assay
Statistical Analysis
-
In the myristoylation-deficient (G2A) reporter pseudovirus system, an equal amount of pGFP-2A-P1-WT or pGFP-2A-P1-G2A, pT7-EV71-luciferase and pT7-RNA-Pol were cotransfected into HEK 293T cells to encapsidate reporter pseudovirus. After 48 h post transfection, the cells containing reporter pseudovirus were lysed for detection of viral protein expression by Western blotting; while the supernatant was collected from the cell culture and concentrated 150-fold for detection of encapsidated virions by Western blotting and observation of virus morphology by TEM. As shown in Fig. 1A, 1B. The viral VP3 protein, a capsid protein of EV71, was detected in the G2A virus group, indicating that G2A protein expression was normal. Furthermore, TEM analyses revealed G2A virus particles were nearly 30 nm in diameter, which was consistent with the shape and size of WT EV71.
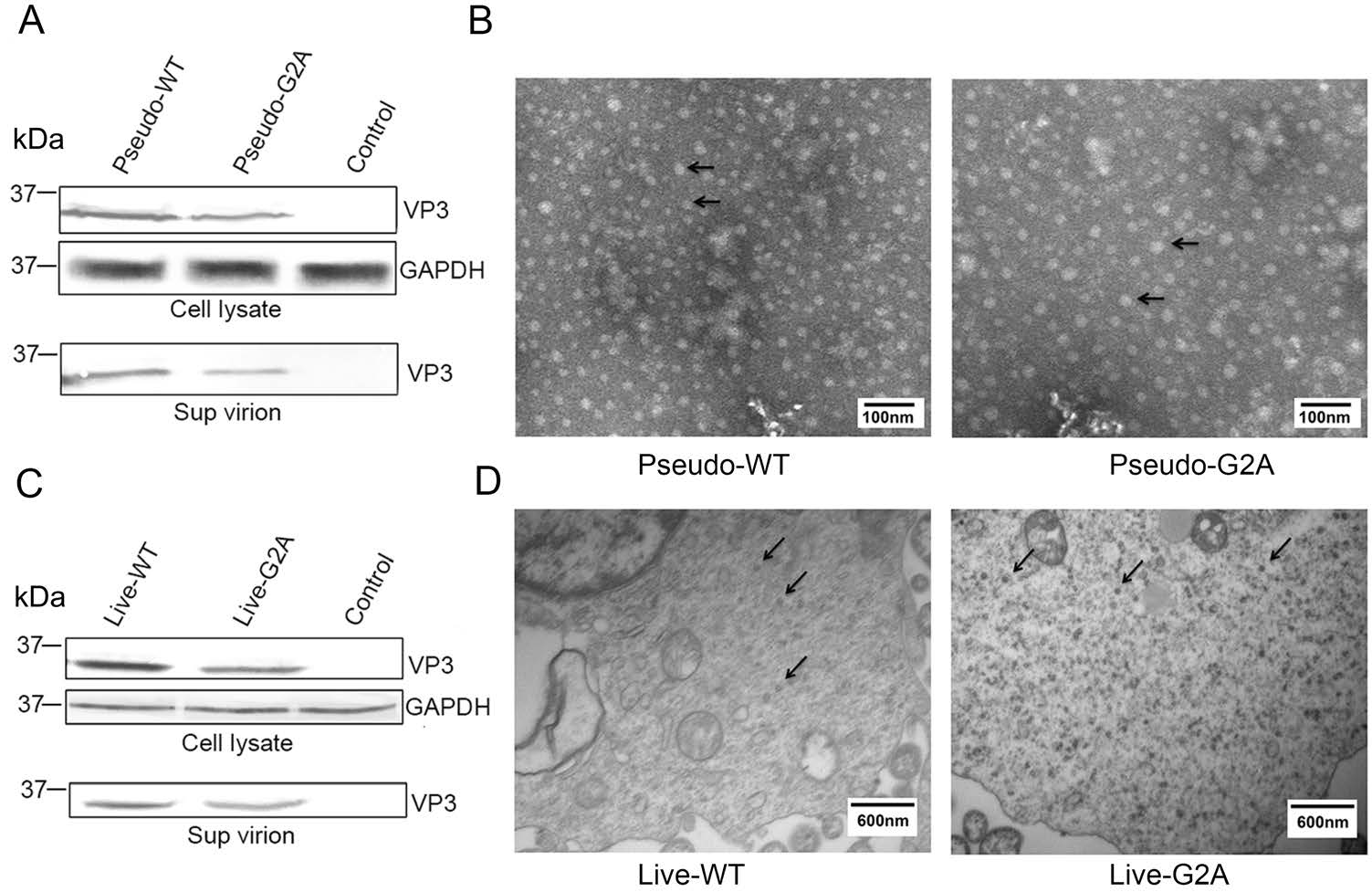
Figure 1. Effect of myristoylation on viral protein expression and morphology of virus particles. A Western blotting analysis of the cell lysate and supernatant of cells transfected with pGFP-2A-P1-WT or pGFP-2A-P1-G2A using rabbit anti-VP3 poly antibody. Sup virion: virions in supernatant. Pseudo-WT: wild type pseudovirus; Pseudo-G2A: myristoylation-deficient pseudoviruses; Control: supernatant of 293T cells transfected with empty vector. GAPDH: internal control. B Transmission electron microscope images of myristoylation and myristoylation-deficient (G2A) reporter pseudoviruses. The arrow points to the virus particles. Scale bar, 100 nm. C Western blotting analysis of the cell lysate and supernatant of cells transfected with pT7-EV71-WT or pT7-EV71-G2A using rabbit anti-VP3 poly antibody. Sup virion: virions in supernatant. Live-WT WT: live myristoylation virus; Live-G2A: live myristoylation-deficient viruses. Control: supernatant of 293T cells transfected with an empty vector. GAPDH: internal control. D High-pressure cryo-electron microscopy images of myristoylation and myristoylation-deficient (G2A) viruses. The arrow points to the virus particles. Scale bar, 600 nm.
In myristoylation-deficient (G2A) virus system, an equal amount of pT7-EV71-WT or pT7-EV71-G2A and pT7-RNA-Pol were co-transfected into HEK 293T cells to encapsidate virus. The supernatants were concentrated 150-fold and used for Western blotting, while the cell lysates were collected for high-pressure cryo-electron microscopy. As shown in Fig. 1C, 1D. The results of Western blot analysis indicated that the viral VP3 protein was expressed in the G2A virus group, indicating that protein expression of G2A virus was normal. Furthermore, Cryo-SEM analyses revealed G2A virus particles were almost 30 nm in diameter and consistent in shape and size compared with WT EV71. From these experimental results, we show that in either virus system, the loss of myristoylation does not affect the normal expression of viral proteins or the morphology of the viral particles.
-
The loss of myristoylation did not affect virus synthesis via plasmid transfection; however, whether it affects the virus in other aspects remained unclear. To further explore the role of myristoylation in EV71, we used the G2A reporter pseudovirus and live virus to infect cells. In the reporter pseudovirus system, WT and G2A reporter pseudovirus, with equal amounts of viral RNA, were used to infect 293S cells (293T cells with stable expression of EV71 receptor SCARB2), and infectivity was determined by measuring luciferase activity. An approximately 18 h post-infection (hpi), the infected cells were collected, and the level of intracellular fluorescent expression was measured. As shown in Fig. 2A, we found that the fluorescent activity decreased to 1/500 in the G2A reporter pseudovirus compared with the pseudo-WT after infection. We subsequently measured the amount of intracellular viral RNA 24 h after infection and compared it with that of pseudoWT. Results showed that the amount of intracellular viral RNA extracted from cells infected with G2A reporter pseudovirus decreased to 1/50 comparing to the pseudoWT (Fig. 2B). We also sought to compare the infectivity of pseudo-WT and pseudo-G2A in 293T cells. However, this proved difficult as 293T cells were resistant to infection by both pseudoviruses (Supplementary Fig. S2).
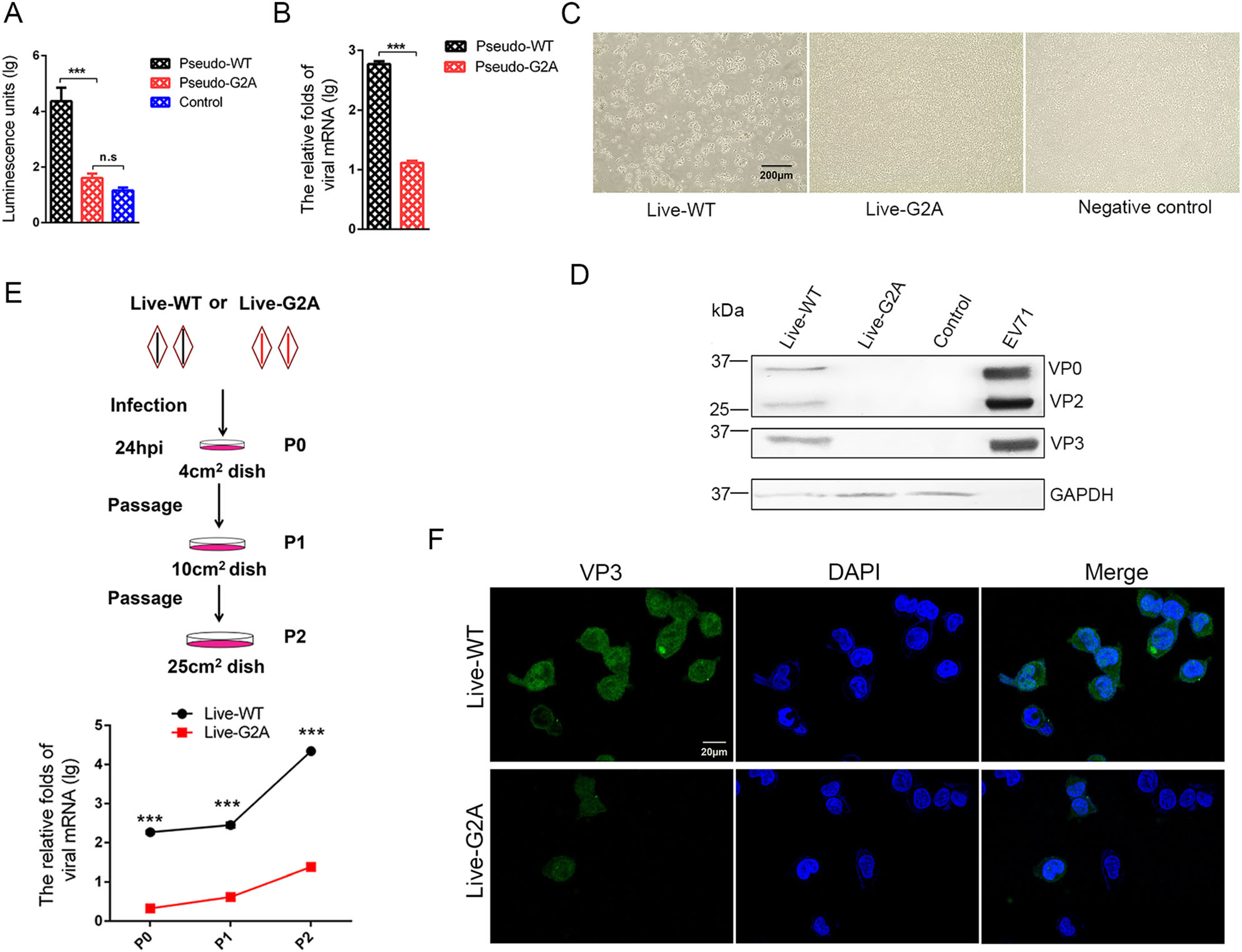
Figure 2. Effect of myristoylation on virus infectivity. A Luciferase activity analysis, B RNA content analysis of myristoylation-deficient (G2A) reporter pseudovirus compared to the Pseudo-WT virus. Myristoylation and myristoylation-deficient (G2A) reporter pseudoviruses with the equal amount viral RNA infected 293S cells, luciferase activity and viral RNA amount were detected 18 h and 24 h respectively after infection. Control represents empty cells that were not infected. Data presented as mean ± SD of three independent experiments. ***P < 0.001. ns non-significant. C Representative photography of cytopathic effects (CPE) analysis of myristoylationdeficient (G2A) virus compared to the WT virus. Myristoylation and myristoylation-deficient (G2A) viruses with the equal amount viral RNA infected 293S cells. At 72 hpi, the cells produced cytopathic effects (CPE) and were photographed and recorded. Live-WT: live myristoylation virus; Live-G2A: live myristoylation-deficient viruses. Scale bar = 200 μm. D Western blot analysis of cells infected with myristoylation-deficient (G2A) virus indicates altered protein expression compared to the WT virus with anti-EV71 VP2/VP3 rabbit polyclonal antibody. GAPDH was the internal control. EV71 inactivated virus was a positive control and 293S was the empty cell control. E The scheme of infected cell blind passages and the measurement of viral RNA in each passage. Myristoylation (WT) and myristoylation-deficient (G2A) viruses with an equal amount of viral RNA infected 293S cells are denoted as P0. After 24 hpi, the cells were passaged twice blindly at a 24 h interval, denoted as P1 and P2. In each passage, living cells were resuspended and transferred to a new, larger dish. Half of the cells in each passage were lysed for intracellular viral RNA extraction and quantification via qRT-PCR. The mRNA levels of viruses were normalized to GAPDH mRNA. Live-WT: live myristoylation virus; Live-G2A: live myristoylationdeficient viruses. ***P < 0.001. F Confocal microscopy images of P2 generation cells infected with WT and G2A virus. WT and G2A live virus were incubated with an anti-EV71 VP3 rabbit polyclonal antibody stained green, and the nuclei were stained blue. Scale bar = 20 μm.
In the myristoylation-deficient (G2A) virus system, WT and G2A live viruses with equal amounts of viral RNA were used to infect 293S cells, and 1 h post-infection, the virus supernatant was discarded and fresh medium was added to the cell culture. Cells were continuously observed and CPE development was noted. As shown in Fig. 2C, the results showed that over time, the live-WT virus group developed cytopathic effects, while no cytopathic effect was observed in the live-G2A virus treatment group. The cells were then harvested for Western blotting analysis, results from which showed that no viral proteins were detected in the live-G2A group compared to the live-WT (Fig. 2D). Based on these results, we speculated that CPE and viral proteins were not detectable in the live-G2A group due to the low infectivity of this virus.
We next performed blind passages of cells infected with WT or G2A live virus, carrying equal amounts of viral RNA, to generate virus accumulation for subsequent detection (Fig. 2E). After 24 h infection (P0), living 293S cells were transferred to new larger culture plates. As the cell culture area expanded, the cells proliferated (P1), meanwhile viruses were permitted to undergo continuous replication. Similar passaging was repeated with a 24 h interval to reach P2 cells. Since high MOI of EV71 virus could cause rapid CPE, we reduced the initial feeder virus concentration used to infect 293S cells to maintain live cells throughout the blind passages. As shown in Fig. 2E, the amount of live-WT viral RNA increased rapidly over time. Although the amount of live-G2A viral RNA also increased, it was significantly lower than that observed for the WT group. Furthermore, immunofluorescence analysis of EV71 VP3 protein level in P2 cells (Fig. 2F) revealed both live-WT and live-G2A viral proteins were observed in cells. The difference was that viral protein expression in live-WT infected cells was pervasive and strong, whereas it was sporadic and weak in live-G2A infected cells. From these results we determined that loss of myristoylation affected viral infection, and impacted viral RNA replication specifically, thereby causing reduced viral infectivity.
-
To determine the cause for the reduced viral RNA replication in G2A live virus, we measured the growth curve of the WT and G2A live viruses over 48 h. WT and G2A viruses carrying an equal amount of viral RNA were used to infect 293S cells, which were then harvested at different time points post-infection to measure the amount of viral RNA produced in the cells. As shown in Fig. 3A, we found that the amount of live-WT viral RNA was significantly higher than that of live-G2A. To support our results, in addition to mutating the site of myristoylation, we also employed the myristic acid inhibitor, 2-Hydroxymyristic acid (2-HMA, Sigma, USA) which competitively blocks covalent attachment of myristic acid to the peptide chain (Corbic Ramljak et al. 2018; Tan et al. 2016) as a positive control for myristoylation inhibition. The 293S cells contained a final concentration of 100 μmol/L 2-HMA and were infected with the live-WT virus. Progeny nonmyristoylated viruses were then collected and used in infection for measurement of growth curves. The single growth curves of the live-WT, live-G2A virus, and 2-HMA treated live-WT virus were measured at indicated time points. As shown in Fig. 3B, we found that the rate of virus replication among the three groups did not differ significantly at 3 hpi. However, from 6 hpi, the replication rate of live-WT increased rapidly; while, the viral RNA of liveG2A and 2-HMA-treated live-WT groups increased by only 1.96 and 2.69 times, respectively, at 12 hpi. These results suggest that loss of myristoylation blocks efficient virus replication.
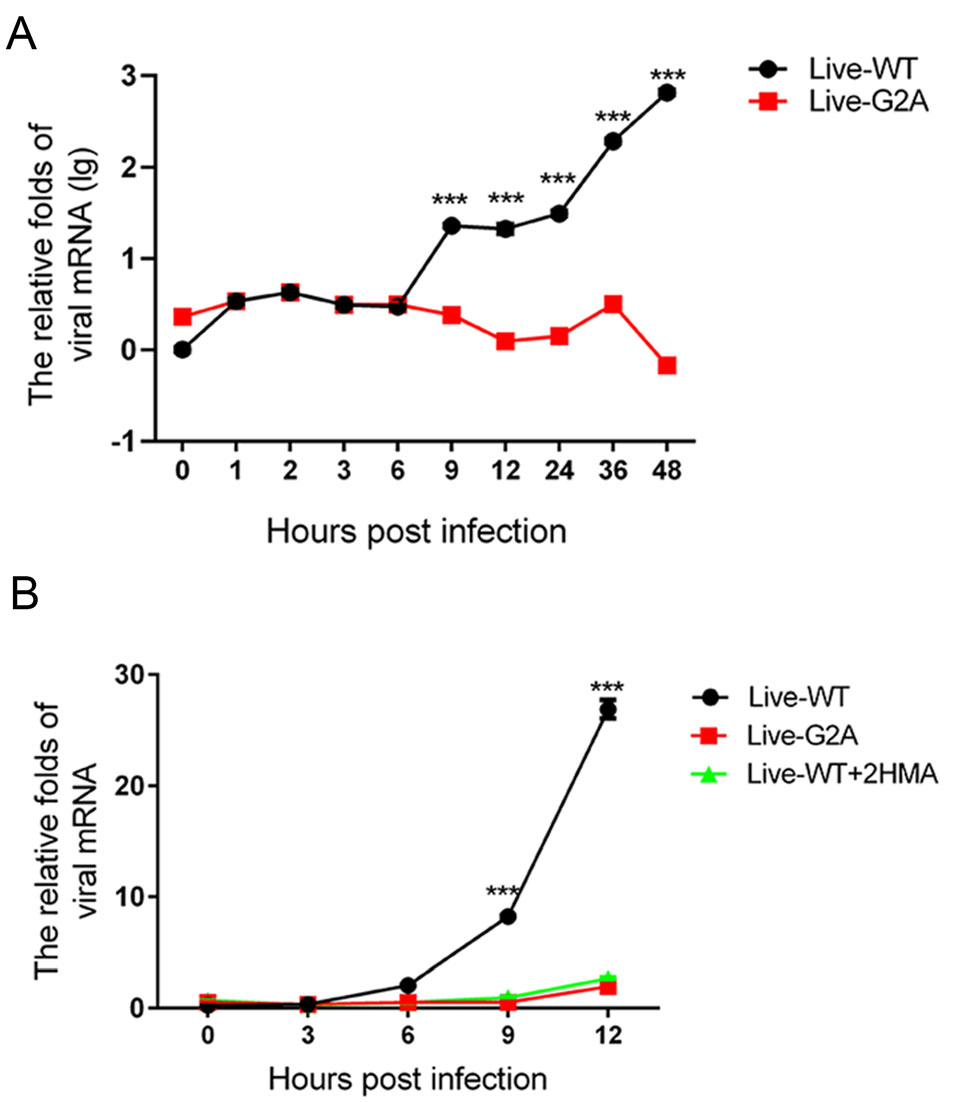
Figure 3. Effect of myristoylation on virus RNA replication. A The growth curve of myristoylation and live G2A viruses. Live WT and G2A viruses containing an equal amount of viral RNA infected 293S cells. Cells were harvested 0, 1, 2, 3, 6, 9, 12, 24, 36, 48 hpi. Cellular RNA was extracted and quantified with real-time PCR. The mRNA levels of viruses were normalized to GAPDH. ***P < 0.001. B The growth curve of WT, G2A live virus, and progeny non-myristoylated viruses. Cells were harvested at indicated times; viral RNA was extracted and quantified with real-time PCR. The mRNA levels of viruses were normalized to GAPDH. Data presented as mean ± SD of three independent experiments. Live-WT: live myristoylation virus; Live-G2A: live myristoylation-deficient viruses. Live-WT + 2 HMA: Progeny non-myristoylated viruses; ***P < 0.001.
-
In addition, compared to the total RNA, the amount of negative-stranded RNA was also measured since during EV71 viral RNA replication, the virus must synthesize negative-stranded RNA using virus mRNA as a template (Paul et al. 2000). To this end, live-WT and G2A viruses carrying an equal amount of viral RNA were used to infect 293S cells with or without 2-HMA. In addition to 2-HMA, we also used another inhibitor, NH4Cl. EV71 viruses enter their host cells by receptor-mediated endocytosis accompanied by destabilization of the endosomal membrane and release of viral genome into the cytoplasm (Lin et al. 2012). Specific compounds, such as NH4Cl (Eash et al. 2004; Olokoltsov et al. 2007), are capable of inhibiting endocytosis. Hence, we chose to include such an inhibitor to verify whether this phenomenon is consequential for EV71 RNA release. We added 40 mmol/L NH4Cl to cells before EV71 infection. As described previously, WT and G2A viruses carrying an equal amount of viral RNA were used to infect 293S cells with or without the two inhibitors, after which we measured the amount of viral negative RNA at different time points. As shown in Fig. 4. We found that the amount of negative RNA for the WT virus increased over time, while that of G2A viruses remained unchanged. Hence, the loss of myristoylation on VP4 appeared to block the generation of negative RNA, with the same result observed in virus-infected cells with inhibitors. According to these results, we conclude that viral RNA was prevented from being released into the cells infected with G2A viruses. The decrease of viral mRNA releasing is seemingly related to the early events of viral infection. Conversely, loss of myristoylation does not affect virus binding to entry into the host. Since the capsid VP4 protein linked to the myristoylation was on the interior of the capsid (Chow et al. 1987; Filman et al. 1989), it would not be capable of directly contacting the host cell surface.
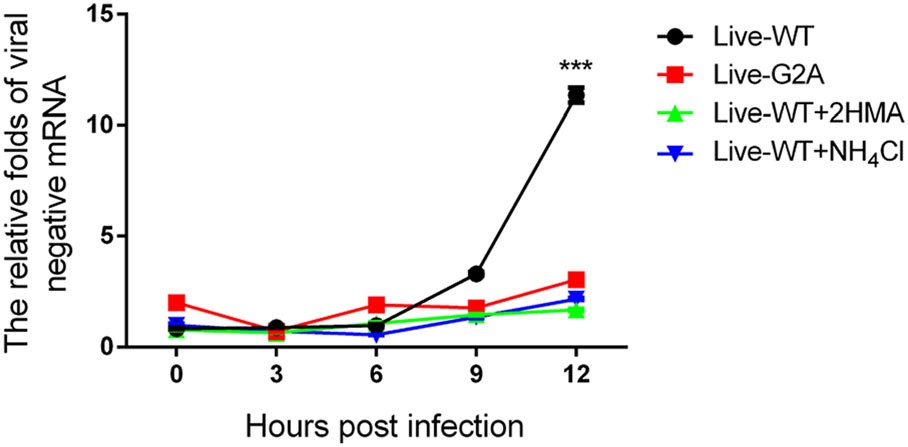
Figure 4. Negative viral RNA content in cells with or without inhibitors infected with WT and G2A live viruses. 293S cells without NH4Cl were infected with live WT, G2A virus and progeny non-myristoylated viruses, and cells with NH4Cl were infected with live WT virus. Cells with or without NH4Cl were harvested at indicated time points. Negative viral RNA content was then measured. The RNA levels of viruses were normalized to GAPDH. Data presented as mean ± SD of three independent experiments. Live-WT: live myristoylation virus; Live-G2A: live myristoylation-deficient viruses. Live-WT + 2 HMA: Progeny viruses without myristoylation, Live-WT + NH4Cl: live myristoylation virus infected cells with inhibitor NH4Cl, ***P < 0.001.
-
It has been clearly reported that myristic acid generally interacts with membrane structures (Wright et al. 2010) as that has been demonstrated in various viruses (Strecker et al. 2006; Wright et al. 2010; Zhu et al. 2017). Hence, we suggested that myristyl groups may also interact with the cell membrane for EV71. To this end, pVP4-WT-GFP or pVP4-G2A-GFP were transfected into 293T cells, protein localization was observed using laser scanning confocal microscopy after 24 h. We found that the localization of VP4-G2A-GFP changed significantly compared to the WT (Fig. 5A). The distribution of WT VP4 proteins followed normal aggregation patterns; while the distribution of proteins in G2A mutants was diffuse throughout the cytoplasm. A certain amount of VP4-G2A-GFP and VP4-WT-GFP proteins were also observed in the nucleus, however, a smaller amount was found in the latter compared to the former. Plasmid-transfected cells were harvested and the cytoplasm and membrane structures were separated for Western blot analysis and determination of VP4 protein proportion in the cytoplasm or membrane based on band grayscale in Western blot. As shown in Fig. 5B, C, 71.6% of VP4-WT-GFP proteins were distributed in membrane structures and 25.9% was distributed in cytoplasm. Conversely, only 19.2% of VP4-G2A-GFP proteins were distributed in the membrane structures, while 66.4% were distributed throughout the cytoplasm. From these results, we determine that myristyl groups interact with host membrane structures.
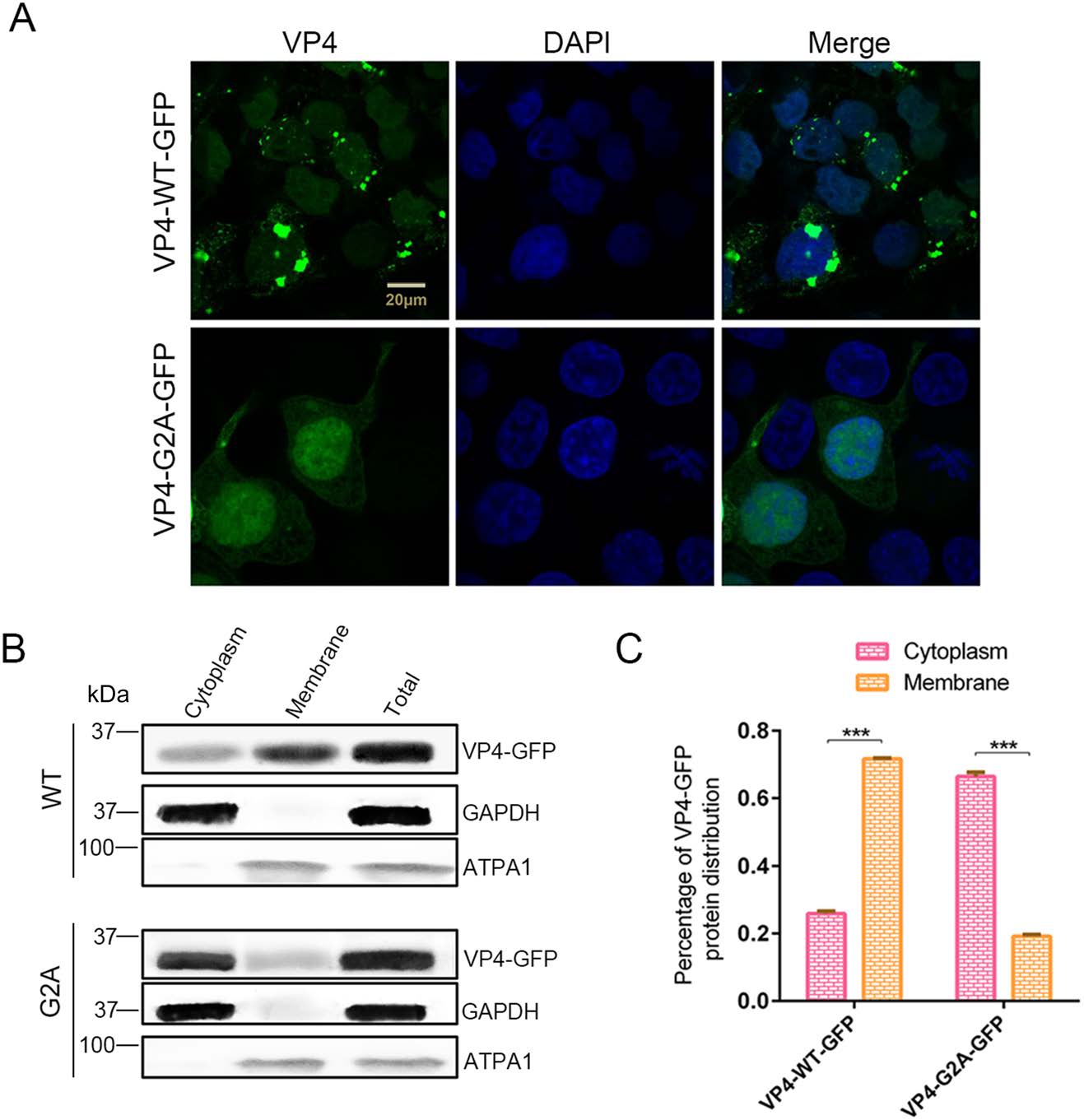
Figure 5. The function of myristoylation of EV71 in membrane interaction. A Confocal microscopy images of VP4-WT-GFP and VP4-G2A-GFP protein distribution. Wild type (WT) VP4 and myristoylation-deficient (G2A) VP4 plasmids were transfected into 293T cells. After 24 h, the distribution of proteins in cells was observed by laser scanning confocal microscopy. The cell nuclei were stained with DAPI (blue) and VP4-WT-GFP or VP4-G2A-GFP is visualized in green. Scale bar = 20 μm. B Western blot analysis of VP4-G2A-GFP protein distribution compared to VP4-WT-GFP. Cytoplasmic fraction and membrane protein fraction were analyzed with Western blotting using an anti-GFP monoclonal antibody. GAPDH was used as an internal control for cytoplasmic protein. ATPA1 was used as an internal control for membrane protein (Zhu et al. 2017). Total is the total proteins from cells transfected with VP4-WT-GFP or VP4-G2A-GFP plasmids. C Percentage of VP4-GFP protein distribution in the cytoplasm or membrane based on grayscale measurement of Western blotting (B). Data presented as mean ± SD of three independent experiments. ***P < 0.001.
-
In human rhinoviruses (HRV) and PV, the interaction of VP4 with membranes following its release from the particle suggests a potential role for VP4 in the membrane alterations required for genome transfer (Davis et al. 2008). Here, we described the EV71 VP4 as synthetic peptides, and investigated its ability to interact with, and induce, permeability in lipid membranes in the form of liposomes. Liposomes containing the carboxyfluorescein (CF) dye were allowed to interact with the WT or G2A VP4 proteins. Since CF becomes quenched at a high concentration, if myristic acid interacts with the liposome, CF will leak out of liposome, lowering its concentration and producing fluorescence. If myristic acid does not interact with the liposomes, CF concentration will remain high in the liposome and not be detected via immunofluorescence. We also performed the same experiment using the truncated VP4 peptide consisting of the initial 20 amino acids at the 5' end of VP4 (VP4-20aa). VP4-20aa included the site of myristoylation, while eliminating the membrane interaction effect of the full VP4 protein (Fuchs and Blaas 2010; Zauner et al. 1995). HRV VP4 was included as a positive control (Zauner et al. 1995). At a concentration of 30 nmol/L, both WT and G2A VP4 induced the release of CF from liposomes. However, the rate of CF release in WT VP4 was faster than that for G2A VP4 (Fig. 6A). After 30 min, the luciferase activity of WT VP4 was distinctly higher than that induced by G2A VP4 (Fig. 6B). However, the rate of CF release from liposomes was not significantly different between VP4-20aa and the full-length VP4, while the rate of CF release induced by unmyristoylated peptides (VP4-G2A-20aa) was slower (Fig. 6C). Finally, the luciferase activity of VP4-WT-20aa was distinctly higher than that induced by the VP4-G2A-20aa (Fig. 6D). From these results, we concluded that both VP4 peptides and short peptides containing 20 amino acids can interact with liposomes so long as they can undergo myristoylation. Hence, myristic acid appears to mediate the viral interaction with the host cell membrane, while myristoylation enhances cell permeability.
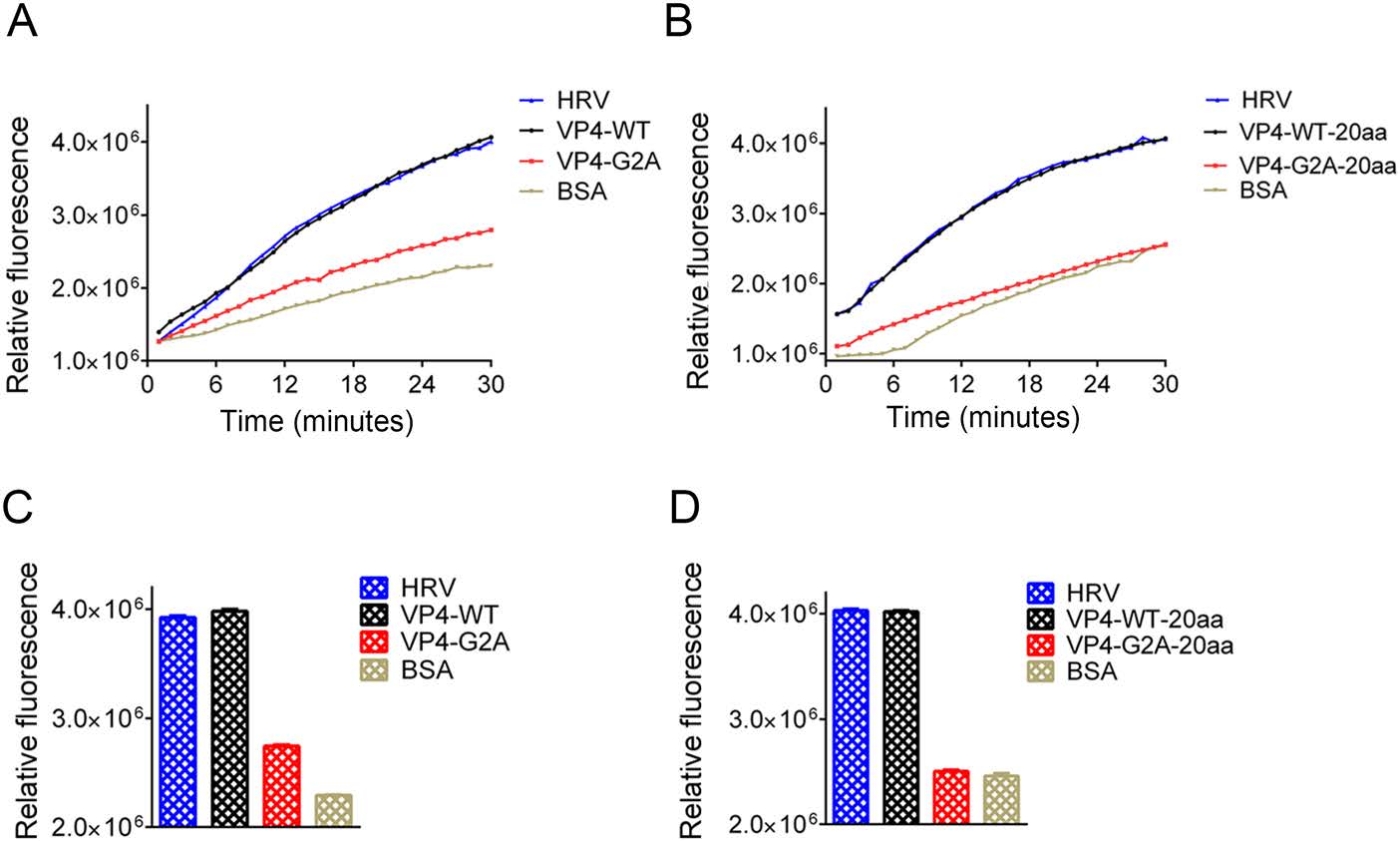
Figure 6. Effect of myristoylation on membrane permeability measured by the release and fluorescent dequenching of carboxyfluorescein (CF) from liposomes. A Luciferase activity was detected after time course of CF dye release induced by 30 nmol/L myristoylated (VP4-WT), unmyristoylated (VP4-G2A) VP4, or BSA alone. The release induced by 30 nmol/L HRV VP4 (HRV) was shown as a positive control. The data shown are representatives of multiple experiments. B Relative rates of CF release for myristoylated VP4 (VP4-WT), unmyristoylated VP4 (VP4-G2A), and BSA alone. Data were calculated by fitting a straight line to the last four data points of the time course. C Luciferase activity was detected after time course of CF release induced by 30 nmol/L myristoylated (VP4-WT-20aa) or unmyristoylated (VP4-G2A-20aa) and BSA alone. The data shown are representative of multiple experiments. D Relative rates of CF release for the first 20 amino acid peptides of myristoylated VP4 (VP4-WT-20aa), unmyristoylated VP4 (VP4-G2A-20aa), and BSA alone, and positive control HRV VP4 (HRV) were calculated by fitting a straight line to the last four data points of the time course. Data are presented as mean ± SD of three independent experiments.
Protein Expression and Virus Morphology are Normal in G2A Virus and G2A Reporter Pseudovirus
G2A Mutation Causes Reduce of Viral Infectivity
Viral Genomic RNA Replication is Blocked in Cells Infected with G2A Viruses
Myristyl Group Affects Early Steps in Viral Infection
The Myristyl Groups Interact with the Host Cell Membrane Structure
The Myristyl Groups Mediate Permeability in Model Membranes
-
Many viruses contain N-terminal modifications to their viral proteins that include glycine linkages with myristic acid, which is a process known as myristoylation. Subsequently, there is a large amount of research on hostdependent viral protein lipid modification. Myristoylation is ubiquitous in viruses and cells and studies have increasingly focused on identifying proteins with new lipid modifications to understand the role of myristoylation in the biological functions of these proteins.
Poliovirus and other members of the picornavirus family were the first non-enveloped viruses identified to contain myristoyl modification on their VP4 capsid protein (Chow et al. 1987). Mutation of the myristoyl site in poliovirus is the main way to study the function of myristoylation. Myristoylation is degraded or completely deleted by substituting a different amino acid residue in the myristoylation consensus sequence (6–8 amino acid residues at the N-terminus of the VP4 protein). In poliovirus, the second glycine at the N-terminus of VP4 was mutated to alanine (Gly-to-Ala) so that VP0 could not be myristoylated, but the replication of mutant RNA was not affected (Krausslich et al. 1990). This indicates that the Gly-to-Ala mutation alone does not affect the function of the viral protein even though myristoylation is impaired. Therefore, in our study, we also performed a Gly-to-Ala mutation on EV71 VP4 to obtain a myristoylation-deficient virus.
Although the myristoylation site has been completely determined, its exact role in the assembly process of the picornavirus is unclear. Moscufo et al. studied the assembly process of the VP4003A.D mutant poliovirus and revealed that its myristoylation level was down-regulated by approximately 50% (Moscufo et al. 1991). Studies have shown that the process of unmyristoylated protomers forming pentamers is attenuated and the unmyristoylated empty capsids accumulate; however, in mature virus particles, the level of myristoylation of the proteins was normal (Krausslich et al. 1990). Therefore, myristoylated protomers are preferentially selected to regulate the kinetics of pentamer formation. Subsequently, during the final stages of viral assembly, including RNA encapsulation and conversion of provirus to mature virions, myristoylated proteins are required to participate in these processes (Moscufo et al. 1991). Experiments have also shown that, contrary to the above results, the processing of myristoylated and non-myristoylated proteins in the mutants is similar (Krausslich et al. 1990). In our study, we demonstrated that the loss of myristoylation did not affect protein expression or the appearance of EV71 virus particles. After transfecting the mutant plasmid into cells, viral proteins were detected in the mutant variant. The appearance of the mutant and WT viruses were indistinguishable by electron microscopy. As shown in Fig. 1.
Although protein production and virus appearance were not affected by the loss of myristoylation, we did observe reduced infectivity in the mutant virus. Poliovirus with the Gly-to-Ala mutation (G4002A) could not infect HeLa cells (Moscufo et al. 1991), and when five amino acids at the N-terminus of poliovirus were mutated, the first (Gly-to-Arg), second (Ala-to-Pro) and fifth (Ser-to-Pro) position mutations prevented myristoylation of the protein precursor P1 and were lethal to the virus (Marc et al. 1989). These findings are consistent with the conclusions in our study, which showed that in pseudoviruses, the loss of myristoylation led to a decrease in viral infectivity (Fig. 2A, 2B). In myristoylation-deficient virus system, we found that CPE could not be produced after the myristoylation-deficient virus had infected cells (Fig. 2C). Viral proteins were also not detected in infected cells (Fig. 2D). Through the process of cell blind passaging, viruses were accumulated and detected in infected cells via immunofluorescence assaying (Fig. 2G), however, viral RNA replication was low (Fig. 2F). Furthermore, we compared the growth curve of G2A to WT (Fig. 3). It is suggested that loss of myristoylation blocked the viral RNA replication, which is consistent with the results reported in common cold virus (Mousnier et al. 2018). Negative-strand RNA was also measured after virus-infected host cells, we found that the negative RNA amount of WT virus increased over time, but the amount of myristoylation-deficient, virus infected the cells with inhibitor 2-HMA and NH4Cl decreased significantly (Fig. 4). Myristoylation probably plays a vital role in the early stage of infection, but may not be involved in receptor binding. Myristyl groups were on the interior of the capsid. Surfaces of major picornavirus have a corrugated topography found at the five-fold axis of symmetry, surrounded by a deep depression (canyon) and another protrusion at the threefold axis. The canyons of enteroviruses are the sites of interaction with cell receptors (Chapman and Rossmann 1993). As shown in Fig. 1, as long as there was viral RNA as a template for viral replication, the process of RNA replication, viral proteins expression, and viral assembly could operate normally in the G2A mutant. During the replication of EV71 viral RNA, the virus needs to synthesize negative-strand RNA using virus mRNA as a template (Paul et al. 2000). According to the results, we conclude that there was no viral RNA releasing into the cells infected with myristoylation-deficient viruses.
There are many N-myristoyl-modified proteins that bind to the membrane (Courtneidge and Bishop 1982; Heuckeroth and Gordon 1989; Johnson et al. 1990; Rein et al. 1986; Rhee and Hunter 1987), and we asked if the distribution of VP4 proteins was associated with its myristoylated or unmyristoylated status after host cell infection. We found that in the cells transfected with myristoylation-deficient VP4, the localization of viral proteins changed from typical aggregation points at the membrane to a more dispersed pattern of localization in the cytoplasm (Fig. 5A). A plasma membrane separation experiment that indicated myristoylation of EV71 does affect virus interaction with the hose cell membrane structure (Fig. 5B, 5C). We found that myristyl groups could interact with the host membrane structure. To more directly illustrate the role of myristoylation of EV71 interactions with membrane structure, we performed a liposome leakage assay. From these results, we found that the myristoylated VP4 protein interacted with the endocytic vesicle-like structure to release CF in it, and the myristoylation-deficient VP4 protein attenuated this effect (Fig. 6A, 6B). After disrupting the integrity of the VP4 protein, the loss of myristoylation significantly diminished the interaction of the VP4 protein with the host cell membrane structure (Fig. 6C, 6D). In enteroviruses, the capsid undergoes a conformational change that allows the viral RNA to be transferred across the plasma membrane and/or endosomal membrane into the cytoplasm through a yet completely defined mechanism (Strauss et al. 2013), and myristoyl group could affect viral RNA release. From these concludes, we speculate that during virus uncoating, myristoyl group interacts with endosomal membrane and disrupts the membrane, allowing viral RNA to be released into the cytoplasm. However, VP4 is only one capsid protein unit of EV71 and does not represent the natural condition of viral infection. Further exploration is needed to understand its mechanism.
Based on the above experimental results, we hypothesize that during the EV71 infection process, especially uncoating, VP4 extends from the interior of the five-fold axis and myristic acid covalently linked to the N-terminus of VP4 that interacts with the membrane of endocytic vesicle, resulting in the broken of the endocytic vesicle. A channel is then formed, and the viral genome leaves the capsid through the channel to enter the host cell.
This work is important to the research field in developing a fundamental understanding of enteroviruses and their life cycles. It also potentially has implications in clinical research or treatment of viral infection.
-
This study was supported by the National Natural Science Foundation of China (Grant No. 31770184).
-
WS, and CJ concepted and supervised the study. JC and MQ performed the experiments. HL, XW, and FL assisted to perform part of the experiments. MQ, AH, YZ, BS, LC, SW and CJ helped to analyze the data. JC, WS and CJ drafted and edited the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: