














HTML
-
Many viruses with circular DNA genomes, including circular replication-associated protein (Rep)-encoding single-stranded (CRESS) DNA viruses, have been reported in both human and animal virome studies (Sikorski et al. 2013a; Tan le et al. 2013; Cui et al. 2017; Kaszab et al. 2018; Kraberger et al. 2018; Varsani and Krupovic 2018). These viruses are typically characterized by a small circular genome (< 10 kb) coated by capsid proteins, without an envelope structure. Eukaryote-infecting CRESS DNA viruses, such as porcine circovirus 2 (PCV2), have mainly been identified within the family Circoviridae (Gillespie et al. 2009; Meng 2013). However, a convincing causal relationship between the animal-associated viruses from other CRESS DNA viral families (e.g., Smacoviridae and Genomoviridae) and any disease, has yet to be documented (de Rezende et al. 2018; Steel et al. 2016; Walters et al. 2017). However, infections with these viruses may play a role in disorders of the host immune system and become an indirect factor for diseases caused by other pathogens (Shulman and Davidson 2017).
According to a recent report issued by the International Committee on Taxonomy of Viruses (ICTV) (https://talk.ictvonline.org/), eukaryote-infecting CRESS DNA viruses have been classified into six families: Bacilladnaviridae, Geminiviridae, Nanoviridae, Genomoviridae, Circoviridae, and Smacoviridae. The viruses of the families Bacilladnaviridae, Geminiviridae, and Nanoviridae have been demonstrated to infect diatoms and plants in previous studies (Tomaru et al. 2013; Heydarnejad et al. 2017; Zerbini et al. 2017), while members of the Genomoviridae, Circoviridae, and Smacoviridae families have been identified in the feces of a variety of vertebrates, including humans and pigs (Li et al. 2010; Krupovic et al. 2016; Rosario et al. 2017; Varsani and Krupovic 2018). There are obvious differences in the aspects of genome characteristics, evolution, and pathogenicity among CRESS DNA viruses (Malathi and Renuka Devi 2019).
In recent years, many unclassified CRESS DNA viruses have been discovered using viral metagenomic methods (Shan et al. 2011; Guo et al. 2018; Rosario et al. 2019; Wang et al. 2019). Fur seal feces associate circular DNA virus (FSfaCV) was first identified in the fecal samples of New Zealand fur seals in 2013 (Sikorski et al. 2013b). The second report of the virus was published in 2017, which reported that FSfaCVs were detected in stool specimens of pigs in Japan (Oba et al. 2017). The FSfaCV genome has two major open reading frames (ORFs) in opposite directions. ORF1 encodes a major structural capsid protein, while ORF2 encodes replication-associated proteins (Sikorski et al. 2013b). No further information about this virus is available.
In this study, we report for the first time the identification of FSfaCVs in pigs in Anhui Province, China. We also preliminarily described the genome characteristics and prevalence of these viruses in this region.
-
A total of 600 nasal swabs were collected from a slaughterhouse during a surveillance of swine influenza in Anhui Province in 2017 (Fig. 1). Each of the nasal swabs was immersed in 1.5 mL sterile phosphate buffer saline (PBS), and half of the solution was used to detect the swine influenza virus. The remaining approximately 0.8 mL was pooled into one sample for virome analysis. Specimen processing and viral nucleic acid library preparation were performed as previously described (Shan et al. 2011). Briefly, the mixed samples were centrifuged at 13, 000 × g for 20 min to remove impurities and filtered through 0.45-μm- and 0.22-μm-filter membranes. The pellet was dipped in PBS overnight after ultracentrifugation at 160, 000 ×g for 4 h at 4 ℃ (Optima XPN-100 Ultracentrifuge, Beckman Coulter, Krefeld, Germany). The precipitates were repeatedly blended and dissolved in PBS. To remove the exogenous nucleic acid contamination, the sample was treated with DNase I and RNase I. Viral DNA/RNA was extracted from samples using EasyPure Viral DNA/RNA kit (TransGen, Beijing, China). Random PCR program was performed as follows. The first strand cDNA was synthesized with random primer of K9N: 5′-GACCATCTAGCGACCTCCCANNNNNNNNN-3′ and PrimeScript II RTase (Takara, Dalian, China) at 42 ℃ for 3 h, and then inactivated at 95 ℃ for 5 min. The second strand cDNA was synthesized with DNA Polymersae I Large (Klenow) Fragment (Promega, Madison, Wisconsin, USA) at 37 ℃ for 3 h, and then inactivated at 75 ℃ for 10 min. The DNA/cDNA was then amplified in a total reaction volume of 50 μL, which included 2 × KOD FX Neo buffer, 0.5 mmol/L each dNTP, 5 μL nucleotide, 10 mmol/L random primer of K9 (GACCATCTAGCGACCTCCCA) and 1 U KOD FX Neo DNA polymerase (Toyobo, Osaka, Japan). Finally, amplification was performed with 1 cycle of 94 ℃ for 2 min, followed by 40 cycles of 10 s at 98 ℃, 30 s at 55 ℃ and 2 min at 68 ℃. The PCR products were assessed by agarose gel electrophoresis. A total weight of 6 μg of random PCR products was submitted to Shanghai Personalbio Company and sequenced by Illumina HiSeq.
Figure 1. Sampling sites located in Anhui Province of China (Lu'an, Anqing, and Chuzhou city). The map was generated using QGIS Desktop 3.4.3 software.
-
Raw read data generated by Illumina sequencing were analyzed on a local viral metagenomic analysis platform. Briefly, the resulting reads were analyzed using the FastQC program (Brown et al. 2017) and the Cutadapt (Martin 2011) software to obtain clean data and were then assembled into contigs using the MEGAHIT software (Li et al. 2016) with default settings. The assembled contigs were noted by BLASTn (Zhang et al. 2000) with a cut-off E-value of 10−5 against a complete genome sequence database of all known viruses. We counted 12 contigs that showed high nucleotide sequence similarities with FSfaCV (Table 1).
Contig no. Length (nt) Description Accession no. E-value Contig 277591 2774 FSfaCV isolate as50 KF246569 0 Contig 430548 2406 FSfaCV isolate as50 KF246569 0 Contig 24164 1041 FSfaCV isolate as50 KF246569 0 Contig 113448 961 FSfaCV isolate as50 KF246569 1.21E−163 Contig 5645 546 FSfaCV isolate as50 KF246569 2.70E−88 Contig 15591 1105 FSfaCV isolate as50 KF246569 2.70E−61 Contig 41875 398 FSfaCV isolate as50 KF246569 7.41E−43 Contig 170989 996 FSfaCV isolate as50 KF246569 3.21E−45 Contig 355689 342 FSfaCV isolate as50 KF246569 1.26E−94 Contig 142897 466 FSfaCV isolate as50 KF246569 1.08E−76 Contig 263359 334 FSfaCV isolate as50 KF246569 1.93E−137 Contig 87283 404 FSfaCV isolate as50 KF246569 6.92E−103 Contig 277591 2774 FSfaCV isolate JPN1 LC133373 0 Contig 430548 2406 FSfaCV isolate JPN1 LC133373 1.10E−172 Contig 24164 1041 FSfaCV isolate JPN1 LC133373 0 Contig 113448 961 FSfaCV isolate JPN1 LC133373 5.68E−157 Contig 5645 546 FSfaCV isolate JPN1 LC133373 5.75E−95 Contig 15591 1105 FSfaCV isolate JPN1 LC133373 1.25E−59 Contig 142897 466 FSfaCV isolate JPN1 LC133373 3.68E−111 Contig 355689 342 FSfaCV isolate JPN1 LC133373 1.27E−89 Contig 263359 334 FSfaCV isolate JPN1 LC133373 9.30E−111 Table 1. Contigs noted to belong to the genome of FSfaCVs.
-
Based on the multiple sequence alignment results, we found that a gap region (approximately 2600–2780 nt) existed in the longest contig (contig 277591) compared to the genome sequence of the FSfaCV isolate as50. Therefore, overlapping PCR was performed to obtain the full-length genome of the virus. Briefly, a pair of primers (FSfaCV-F: 5′-CCAGTATGTTTTCCGATTG-3′; FSfaCV-R: 5′-CGCTTGTCCCTTATGTCTT-3′) were designed to amplify a 585-bp-long segment (containing part of the Rep and 5′-end intergenic region) in the FSfaCV genome, which completely covered the missing sequence of the contigs. The reaction was carried out in a 50-μL mixture including 25 μL of 2 × Taq PCR StarMix (GenStar), 2 μL of each primer (10 μmol/L), 2 μL of DNA template, and 19 μL of sterile water. The optimal PCR amplification procedure consisted of pre-denaturation at 95 ℃ for 2 min, followed by 40 cycles of denaturation at 98 ℃ for 10 s, annealing at 55 ℃ for 30 s, and extension at 72 ℃ for 40 s, followed by a final extension at 72 ℃ for 10 min. The PCR products were subjected to agarose gel electrophoresis (1.2%) in TAE buffer. The full-length genome sequence obtained was designated as FSfaCV-CHN. The genome sequence was deposited in the GenBank database under the accession number MK462122.
-
To confirm the viral metagenomic analysis results, a retrospective survey of FSfaCV prevalence in pigs in the Anhui Province was conducted. A total of 197 samples, consisting of 79 serum samples, 88 nasal swab samples, and 30 tissue samples (mixture of lymph node and spleen) were collected from clinically healthy pigs in three cities (Lu'an, Anqing, and Chuzhou) (Fig. 1). The details of the samples are shown in Table 2. The viral DNA was extracted from the samples using EasyPure Viral DNA/RNA kit (TransGen, Beijing). Gene segments of FSfaCV were then detected using the above-mentioned overlapping PCR method. Briefly, the reaction was carried out in a 20-μL mixture including 10 μL of 2 × Taq PCR StarMix (GenStar), 1 μL of each primer (10 μmol/L), 1 μL of DNA template, and 6 μL of sterile water. The optimal PCR amplification procedure was performed with three-step cycles as mentioned above. The PCR products were subjected to agarose gel electrophoresis (1.2%) in TAE buffer. From the FSfaCV-positive samples, 11 gene segments were cloned into the pMD18-T vector. These included seven serum samples—collected from Lu'an and Anqing—two nasal swab samples—collected from Lu'an—and two tissue samples collected from Chuzhou. Each ligation mixture was transformed into Escherichia coli DH5α competent cells and positive clones, with appropriate inserts being screened by colony PCR and subsequently sequenced by Kumei Comate Bioscience Co., Ltd. All of the gene fragments generated in this study were deposited in the GenBank database under the accession numbers MK462123–MK462133.
City location Source Sample type Prevalence (%, positive/total) Lu'an Slaughterhouse A Serum 30.0 (15/50) Nasal swab 52.3 (46/88) Anqing Farm A Serum 100.0 (20/20) Farm B Serum 100.0 (9/9) Chuzhou Slaughterhouse B Lymph node and spleen 81.3 (13/16) Slaughterhouse C Lymph node and spleen 57.1 (8/14) Total 56.4 (111/197) Table 2. The FSfaCV prevalence in pigs in 2017 of Anhui Province.
-
The genome organization of the FSfaCV-CHN was generated using SeqBuilder software in the DNASTAR package (DNASTAR, Madison, WI, USA). The stem-loop structures were detected using the DNA folding prediction software Mfold (Zuker 2003). Open reading frames (ORFs) in the full genome of FSfaCV were identified using ORFfinder (http://www.ncbi.nlm.nih.gov/orffinder).
-
To learn the phylogeny of the FSfaCVs obtained in this study, a multiple sequence alignment was performed with the amino acid sequence of the Rep of the new virus and reference strains, including FSfaCV-as50 (Sikorski et al. 2013b), FSfaCV-JPN1 (Oba et al. 2017), and 30 representative strains of the families Circoviridae, Smacoviridae, Genomoviridae, Bacilladnaviridae, Geminiviridae, and Nanoviridae. Maximum-likelihood trees based on Rep were then constructed with the default setting of the method. To explore the gene variations of the Chinese FSfaCVs, a neighbor-joining tree based on the 585-bp-long gene segments from the 11 positive samples obtained in this study and that of the FSfaCV isolates CHN, as50 (Sikorski et al. 2013b) and JPN1 (Oba et al. 2017) were also constructed. The methods used for constructing the phylogenetic trees were implemented in MEGA 6.06 (Tamura et al. 2013). The tree topologies were evaluated using 1000 bootstrap analyses.
Sample Collection, Preparation, and Sequencing
Viral Metagenomic Analysis
Determination of the full-length genome of FSfaCV
Survey of FSfaCV Prevalence in Anhui Province
Genome Organization of FSfaCV-CHN
Phylogenetic Analysis
-
Based on the results of the viral metagenomic analysis, 742, 196 reads and 307 contigs were identified as belonging to viral genomes. Of these contigs, 12 showed very high nucleotide sequence similarities with the genome sequences of FSfaCVs evaluated using BLASTn with a cut-off E-value of 10−5 (Table 1). We then obtained the full-length genome sequence of a new virus based on the longest contig by using an overlapping PCR method. This genome sequence is 2915 nt in length, similar to that of the FSfaCV isolates JPN1 (LC133373) (2916 nt) and as50 (KF246569) (2925 nt). The genome-wide pairwise identities between the new virus and FSfaCV-as50 or FSfaCV-JPN1 were 91.3% and 90.9%, respectively. The genomic sequence alignment results of the three FSfaCVs are shown in Supplementary Figure S1. The amino acid length of the Rep (352 aa) of the Chinese strain is consistent with the other two strains, but the length of the Cap (375 aa) of the Chinese strain is slightly shorter than that of the other two strains (397 aa). The number of bases in the stem-loop structure at the 5′ UTR of the Chinese strain is the same as in the other two FSfaCVs. This result indicates that this genome sequence belongs to a fur seal feces-associated circular DNA virus, which we designated as FSfaCV-CHN.
-
Based on the viral metagenomic analysis results, we conducted a retrospective survey of FSfaCV prevalence in pigs in Anhui Province. A total of 197 different samples were collected in November 2018. Using the PCR method developed in this study, we detected a total of 111 samples that were positive for the gene segment, yielding an overall prevalence of 56.4% (111/197), which was lower than the previously reported percentage for the virus in piglets in Japan (Oba et al. 2017). The positive rates of FSfaCVs in different samples—such as serum, nasal swab, or tissue—were variable. It was striking that sera from two farms in Anqing city were all positive (100%) for the virus. The details of the samples and the detection results are shown in Table 2. These results indicate that FSfaCVs have been circulating with a notably high prevalence in the pig population in Anhui.
-
The FSfaCV-CHN genome contains two ORFs in opposite directions (Fig. 2). The ORF1 of FSfaCV-CHN is 1125 nt (81–1205) in length, encoding the capsid protein, while the replication-associated protein (Rep)-encoding gene ORF2 is 1056 nt (2470–1415) in length. Furthermore, the lengths of the intergenic regions between the 5′-ends of the Rep and Cap genes of the FSfaCVs are approximately 470 nt, and the 3′ intergenic regions between the stop codons of two major ORFs of the FSfaCVs are approximately 200 nt. A conserved nonanucleotide motif (5′-TAGTATTAC-3′) was also observed in the FSfaCVs and is located at the apex of the potential stem-loop structure. The Rep gene has been demonstrated to be able to introduce a breach to cleave the viral strand between positions 7 and 8 within the conserved nonanucleotide motif, probably initiating circovirus genome replication via rolling circle replication (RCR) (Rosario et al. 2017). These findings show that the genomic organization of FSfaCV-CHN is similar to that of the two previously identified FSfaCVs.
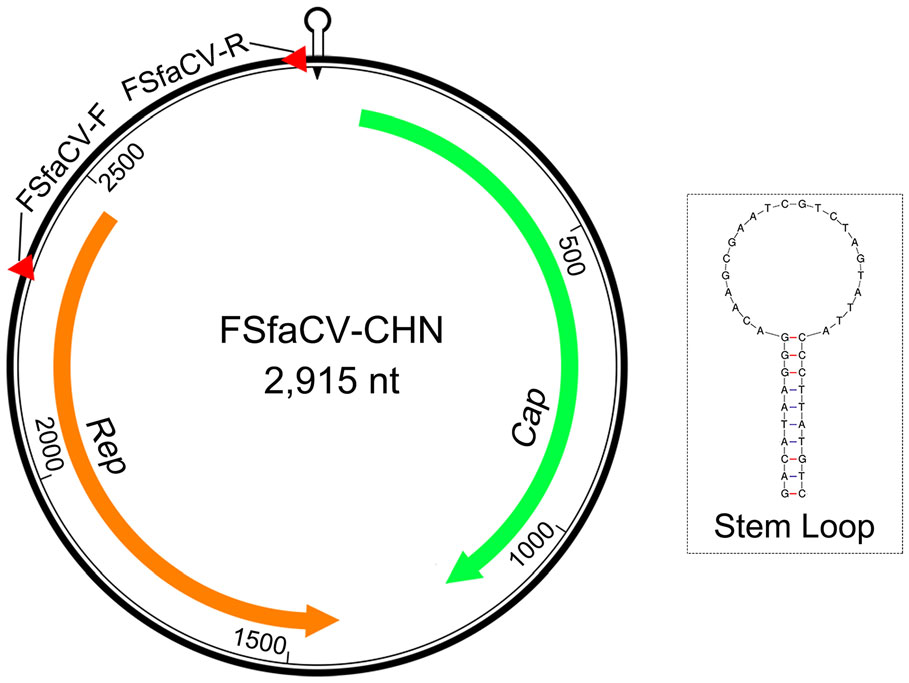
Figure 2. Genome organization of FSfaCV-CHN. Cap indicated by green arrow; Rep indicated by orange arrow. The positions and directions of the primers are marked by red triangles.
-
To further explore the phylogeny of the Chinese FSfaCV isolates, we constructed maximum likelihood trees based on the amino acid sequence of the Rep of FSfaCV-CHN and the reference strains. The three FSfaCVs were located in an independent clade in the Rep tree and were genetically closer to the families Circoviridae and Bacilladnaviridae, suggesting that they might have a common origin in phylogenetic evolution (Fig. 3). All of those FSfaCVs' Rep shared < 55% amino acid identities with the members of other families (e.g., Circoviridae) (Rosario et al. 2017). These findings suggest that FSfaCVs may represent a novel family of CRESS DNA viruses.
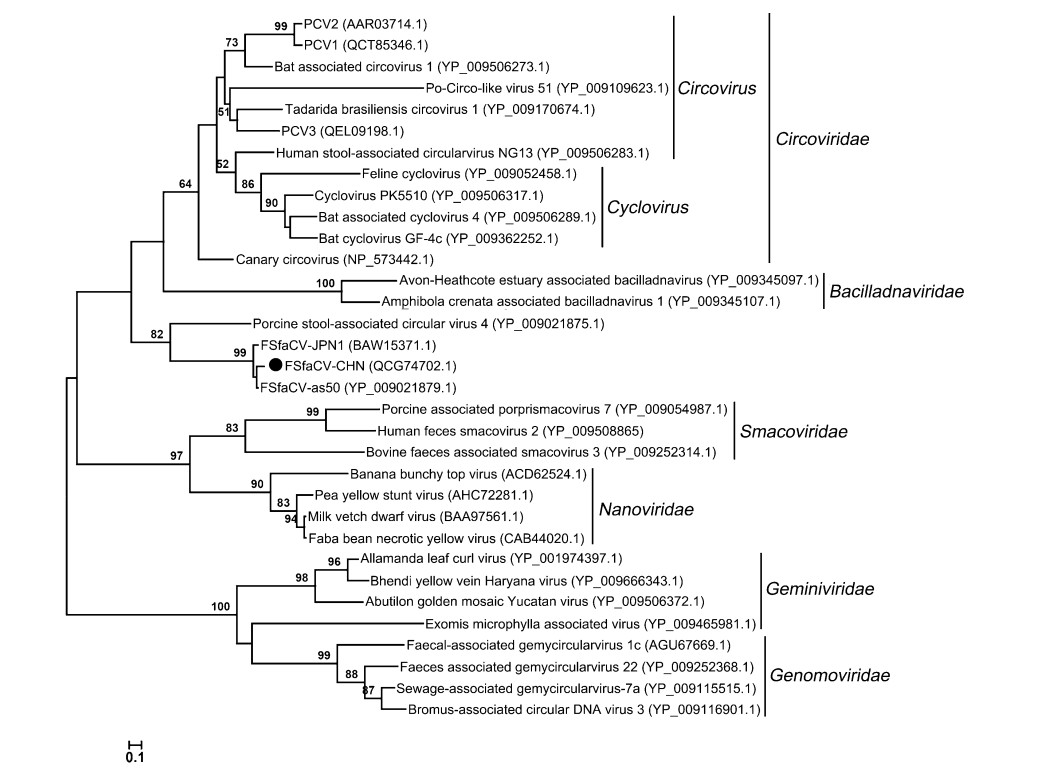
Figure 3. Maximum-Likelihood tree based on the Rep amino acid of CRESS DNA viruses. The tree was constructed using the Maximum-Likelihood method implemented in MEGA 6.06 with bootstrap tests of 1000 replicates. Viruses identified in this study are indicated with black dots. Sequences are labelled with virus names and amino acid accession numbers.
To understand the potential gene variations among Chinese FSfaCVs, we sequenced gene fragments from 11 positive samples, and a neighbor-joining tree was built based on them and on sequences of the reference strains, including that of FSfaCV-CHN, FSfaCV-as50, and FSfaCV-JPN1. As indicated by the tree, all the Chinese viruses clustered into one branch but showed certain phylogenetic variations (Fig. 4). The pairwise nucleotide sequence identities of this gene fragment among all FSfaCVs were 88.7%–99.7%, which were 91.3%–99.7% among the Chinese strains. The results suggest that FSfaCVs in different geographical locations may have independent evolutionary traces.
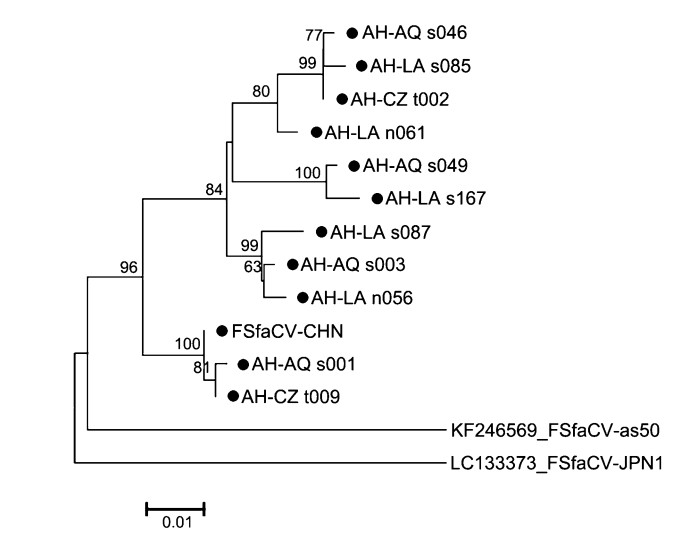
Figure 4. Phylogenetic tree based on 585-bp-long fragments (containing part of the Rep and 5′-end intergenic region) of the FSfaCVs. The tree was constructed using the neighbour-joining method implemented in MEGA 6.06 with bootstrap tests of 1000 replicates. Viruses identified in this study are indicated by black dots. All identified viruses except FSfaCV-CHN strain in this study come from the 111 positive samples. The virus name is composed of Province name (AH: Anhui), city name (AQ: Anqing; LA: Lu'an; CZ: Chuzhou), sample type (s: serum; n: nasal swab; t: tissue) and sample number.
Identification of FSfaCV-CHN
Prevalence of FSfaCV in Anhui Province
Genomic Structure of FSfaCV-CHN
Genetic Evolution of FSfaCVs
-
CRESS DNA viruses are extremely diverse and widespread. They are "key players in the global virome" (Malathi and Renuka Devi 2019). However, their ecological or host impact remains largely unknown (Dayaram et al. 2015; Phan et al. 2016; Kraberger et al. 2018; Zhao et al. 2019). CRESS DNA viruses have been identified in various hosts, including bovines (Li et al. 2011), chimpanzees (Li et al. 2010), bats (Wu et al. 2016), chickens (Lima et al. 2017), humans (Phan et al. 2014; Phan et al. 2016; Cui et al. 2017), and pigs (Stenzel and Koncicki 2017; Zheng et al. 2017). As various new CRESS DNA viruses have been identified in pigs, our understanding of the diversity of these viruses has been enhanced. Some CRESS DNA viruses are important pathogens in pigs, such as porcine circovirus 2 (PCV2), which has been shown to be a potential pathogen of several syndromes known as porcine circovirus-associated disease, causing multisystemic infections in weaned and fattening pigs (Jiang et al. 2017). Therefore, the identification and characterization of novel CRESS DNA viruses is of great importance for evaluating the potential impacts of viruses on the pig industry.
The newly identified FSfaCVs in China showed similar genetic characteristics to the previously identified FSfaCVs. Compared to previous findings, the intergenic regions and the stem-loop structures showed significant differences between the FSfaCVs and other associated circular single-stranded DNA viruses (Krupovic et al. 2016; Rosario et al. 2017; Varsani and Krupovic 2018). These differences might be important evidence for distinguishing FSfaCVs from other associated-circular single-stranded DNA viruses. Furthermore, Rep gene is considered the only phylogenetic marker for CRESS DNA viruses and is widely used for phylogenetic analysis and high-level classification (Simmonds et al. 2017; Zhao et al. 2019). Based on the results of a phylogenetic analysis in which FSfaCVs were clustered together with a stool-associated circular virus within an independent clade on the phylogenetic tree and the identities of the amino acid sequence of the Rep gene between these FSfaCVs and the reference CRESS DNA viruses were all less than 55%. These findings suggest that a new family may be proposed to include FSfaCVs and other similar viruses.
FSfaCVs may infect multiple organs of pigs. In addition to previous studies that identified FSfaCVs from the feces of fur seals (Sikorski et al. 2013b) and from the feces and blood of healthy pigs (Oba et al. 2017), we detected FSfaCVs from nasal swabs, sera, and tissue specimens in healthy pigs, indicating that these viruses were distributed in multiple organs of pigs and that the viruses could also be excreted through respiratory droplets. These findings expand the body of knowledge concerning the biology and epidemiology of the virus. Furthermore, given that the virus was identified from healthy pigs in this study, we assume that this virus may be a component of the common virome community of pigs and may not be a direct pathogen, similar to a previous study on human anelloviruses (Maggi and Bendinelli 2010). Other studies, however, have shown that viruses without direct pathogenicity may weaken host immune function and trigger the infection of other pathogenic agents (Shulman and Davidson 2017).
To date, FSfaCVs have been identified in New Zealand, Japan, and China—regions that are geographically isolated from one another. The viruses identified in these regions have also shown unique genetic characteristics, which suggests that FSfaCVs may be distributed broadly around the world. In this study, an overall positive rate of 56.4% for the virus was detected in pigs in Anhui, China, and a 100% positive rate was found in two farms, suggesting that FSfaCVs might have been circulating with notably high prevalence in the pig population in Anhui. The full-length genome sequences of these 111 positive samples were not obtained, probably because of either the low virus loading in these samples or the amplification strategies we adopted. We are continuing to optimize the reaction conditions to obtain the genome sequences from these positive samples and other samples we collected more recently. Furthermore, although all of the viruses to date have been identified from clinically healthy pigs, given that the novel biological characteristics of these viruses and their pathogenicity in animals remain largely unknown, further research is needed to address the potential risks that these viruses pose to pigs in this region and in other places in China.
In summary, we have described the first identification, full genomic characterization, and preliminary epidemiological investigation of FSfaCVs in pigs in China.
-
This study was supported by the National Key Research and Development Program of China (2016YFD0500705) and Central Public-interest Scientific Institution Basal Research Fund (No. 1610302019002).
-
JW designed the study. ZS and CL performed the experiments. HY, YC and HL collected the samples. LW, ZL, YJ and XH provided experimental support. JW, ZS and CL analyzed the data and drafted the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
All institutional and national guidelines for the care and use of animals were followed and clarified in the article.
-
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link tothe CreativeCommons licence, and indicate if changes weremade. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.

 DownLoad:
DownLoad: