











-
Herpesviridae is a family of enveloped, doublestranded DNA viruses with relatively large complex genomes. They have a wide range of vertebrate hosts, including eight varieties isolated in human, several each in horses, cattle, mice, pigs, chickens, turtles, lizards, fish, and even in some invertebrates, such as oysters (1, 3). The origins of herpesviruses are the subject of interesting but largely unverifiable speculations. The presence of homologs in many viruses indicates that herpesviruses acquired many of their genes from a common ancestral virus or cellular genomes.
The proteins encoded by β-gene including UL2 and UL23 are expressed after the activation of immediateearly proteins (14, 15, 16). They play a critical role in replication stage of DNA. Since all viruses are parasitic to cellular organisms and require molecular machinery produced by their cellular hosts in order to complete their life cycles (4, 5, 7), a homology analysis on the protein conserved among the herpesviruses could provide some clarification of the replication pattern of HSV. Very few papers describe the evolution of early proteins in these pathogens, and the aim of this study is to align selected gene sequences of several early proteins and to perform phylogenetic studies to further clarify the properties of these pathogens.
HTML
-
We used BlastP with an E-value of 10-20 to search all predicted proteins from the set of Herpesvirus or non-virus genomes against the following sequences of HSV1: complete protein translations (proteome) for UL2 and UL23. The GenBank accession numbers of the protein sequences derived in this study are listed in Table 1. Phylogenetic trees were constructed for each protein family present in all species of Herpesvirus analyzed.

Table 1. The GenBank accession numbers of the protein sequences enrolled
-
The sequence data were aligned using MEGA package (version 3.1) (10), and phylogenetic trees were constructed using the neighbor-joining program, from a distance matrix corrected for nucleotide substitutions by the Kimura two-parameter model. Tree stability was assessed via bootstrapping over 500 replicates and if the bootstrap value exceeded 95%, then the cluster was regarded as strongly supported.
UL2 and UL23 encoding protein homologs shared with herpesvirus and other species
Phylogenetic analysis
-
In the UL2 family, homologs were found in 10 species of Herpesvirus and 4 species of bacteria. The sequence from bacteria did not cluster with the herpesvirus sequences. Although the rooting of the tree was not certain, there were two major clusters separated by a significant internal branch. One cluster included uracil-DNA glycosylase of proeukaryotes; the other cluster included UL2 of herpesvirus exclusively from any other virus (Fig. 1). This NJ tree was supported by strong bootstrap value ( > 95%).
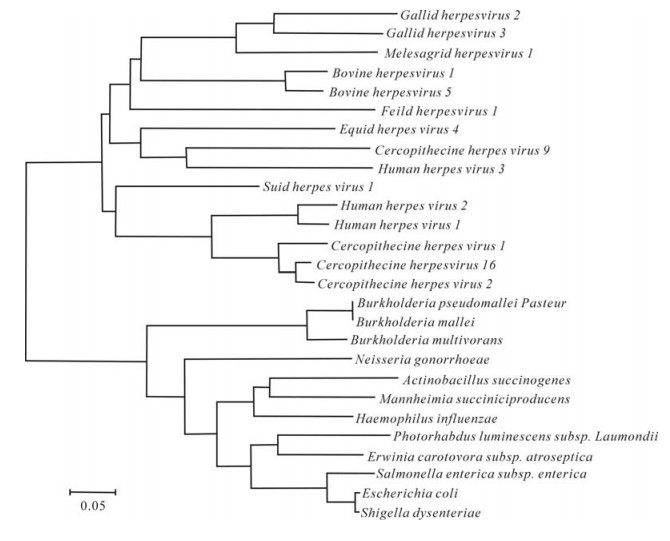
Figure 1. Phylogenetic analysis of UL2 encoding protein homologue of Herpesviridae. A phylogenetic tree was constructed by the neighbor-joining method. Evolutionary distances are to the scale shown.
-
In the phylogeny of Thymidine kinase, there were three major clusters, separated by a highly significant internal branch (Fig. 2). Supported by strong bootstrap value ( > 95%), the tree demonstrated the thymidine kinase and its homologs distributed widely within Herpesvirida. The tree also shows some host specific clustering, with several avian herpesviruses clustered in the first branch. In second cluster, human herpesvirus 1 and 2 are homologous with the Alphaherpesvirinae from other mammalian hosts.
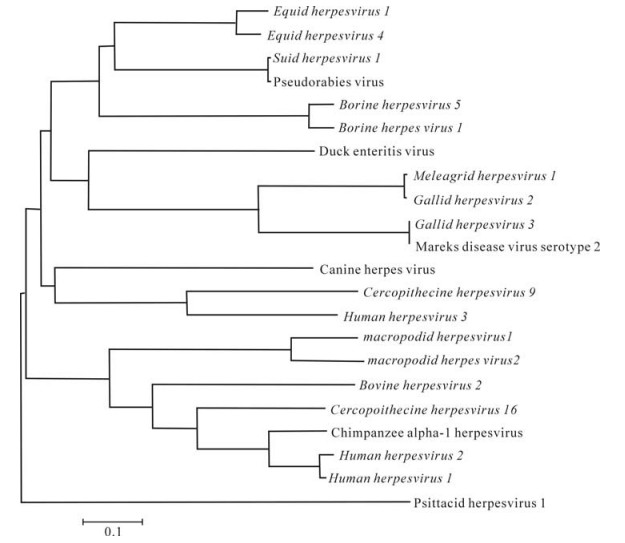
Figure 2. Phylogenetic analysis of UL23 encoding protein homologue of Herpesviridae. A phylogenetic tree was constructed by the neighbor-joining method. Evolutionary distances are to the scale shown.
In total, 2 complete sequences including UL2 and UL23 were selected for different species. These sequences showed 10 to 95% identity to HSV1 sequences. Phylogeny studies using both distance and discrete methods yielded similar results (data not shown), with no obvious geographical or host specificity grouping of isolates evident.
Homologic analysis of uracil-DNA glycosylase encoded by UL2gene
Homologic analysis of thymidine kinase encoded by UL23 gene
-
The origins of herpesviruses is a topic of great interest, and currently of many unverifiable speculations. The similarities in the sequence structure of herpesviruses suggest that they have arisen from a common ancestor. The ancestral virus evolved into at least two highly diverged lineages, one represented by the alpha-, beta-, and gamma-herpesvirus subfamilies that infect birds and mammals, the other represented by the herpesviruses of poikilothermic animals (2, 12, 17).
The increasing prevalence of herpes and corresponding rise of neonatal infection and the implication of Epstein-Barr virus (HHV-4) and Karposi's sarcoma herpesvirus as cofactors in human cancers emphasize the urgent need for a better understanding of this complex virus family (6, 8, 9). In the survey of the β gene product including the UL2 and UL23 proteins of Herpesvirus showed phylogenetic tree topologies indicative of horizontal gene transfer. In the case of UL2, the phylogenetic tree showed three clusters, each of which contained sequences from herpesvirus or bacteria organisms. Theoretically, this topology is consistent with the hypothesis that horizontal transfer occurred from herpesviruses to cellular organisms. This also may suggest ancestral recombination events between these different members. These genes seem to be conserved in different species, which may indicate their common functions in the life cycle of this family of viruses. In the analysis on UL23 protein homologs, there are six conserved regions among Herpesviridae thymidine kinase encoded by UL23. Although the roles of these motifs in protein function are not know, the clusters containing UL23 from Alphaherpesvirinae to Gammaherpesvirinae suggest that this family plays an important role in the herpesviruses life-cycle. Typically, human herpesvirus 1 and 2 are homologous with Herpesvirinae from other mammalian hosts, which suggests the thymidine kinase is related to the ability to establish a lifelong latent infection within sensory neurones of the peripheral nervous system. This deduction is also implied by the evidence of HHV3, also as one member of Alphaher-pesvirinae, which falls outside the herpesvirus 1 and 2 clusters. Thus, in addition to adaptation to specific cellular niches, many of the genes must also function in optimal balance between virus and stimulation of host responses (11, 13, 18).
In summary, phylogenetic analyses has provided evidence that some β-gene products, such as UL2 and UL23 from HSV1, have their homologous genes in their family, and also exist in prokaryotic organisms, indicating that these viruses appear to have been assembled over evolutionary time by numerous independent events of horizontal gene transfer.

 DownLoad:
DownLoad: