











-
Pseudorabies virus (PRV), an important pathogen causing Aujeszky's disease in swine and leading to latent infection (6), is a member of the Alphaherpesvirinae subfamily and could served as a useful model organism for the study of herpesvirus biology (9). Like most of the herpesviruses, PRV has a very large double stranded DNA genome (~143kb) with more than 70 genes identified (3). Functional characterization of these viral genes, by generating null mutants and investigating the resulting changes in phenotype, is very important in understanding the molecular aspects of herpesvirus replication and pathogenesis. For some key genes, smaller targeted mutations are required to identify the distinct functional domains within the proteins (9).
Such studies often require establishment of large numbers of recombinant viruses which are usually created by homologous recombination in infected cells relying on the cellular recombination and repair machinery. However, this can be a laborious and sometimes impossible task, especially if the mutant has a severe growth disadvantage compared to the wild-type virus. Bacterial artificial chromosomes (BACs), single copy F-factor-based plasmid vectors of intermediate insert capacity (15), have now enabled the cloning of complete herpesvirus genomes and infectious virus genomes can be shuttled between Escherichia coli (E. coli) and eukaryotic cells. While herpesvirus BAC DNA engineering in E. coli requires neither restriction sites nor cloning steps and allows the introduction of a wide variety of DNA modifcations, the large size of these bacmids precludes the use of rapid in vitro methods of manipulation commonly employed for construction of small plasmids.
Tn7, a site-specific transposon, transposes almost exclusively to a distinct attachment site named attTn7 within the E. coli genome (1). By introducing this attTn7 sequence into a BAC, Tn7 can serve as an insertion vehicle (4). This is particularly useful if numerous genes and constructs need to be tested for their expression in the context of viral genome. Tn7-mediated transposition has been well exploited for research on functional genomics of baculoviruses (4). Recently, this technology has been applied to bacmid-cloned cytomegalovirus (CMV), a member of the Gammaherpesvirinae subfamily, for rapid recombinant virus construction (2).
In this paper, we report the development of a technology employing Tn7-mediated transposition as a rapid and reliable method for recombinant PRV construction. A lacZα-mini-attTn7 region was inserted into the intergenic region between the gG and gD genes in an attempt to maintain every gene and element of the parental virus. Then green fluorescent protein (GFP) gene was introduced to test the utility of this transposition system and the stability of mini-Tn7 insertions in cell culture. The technology should greatly facilitate the detailed mutagenic studies of PRV.
HTML
-
Plasmids pGS284 and pBecker3, strains GS500 and S17λπ were provided by Lynn W. Enquist, Princeton University, USA. The pGEM-T Easy was purchased from Promega Co. (Maddison, USA), and plasmid pEGFP-N1 from Clonetech Laboratories, Inc. (Mountain View, USA). DNA restriction enzymes, T4 DNA ligase, alkaline phosphatase, exTaq hot start DNA polymerase and 2×GC buffer I were the products of TaKaRa Biotechnology Co., Ltd. (Dalian, China). Plasmids pFBCMV-GFP and pZFBΔtk were constructed and stored in our lab. Strains DH5α and DH10B were stored in our lab. Lipofectin reagent and Delbecco's Modified Essential Medium (DMEM) were purchased from Invitrogen Co. (Carlsbad, USA), and fetal bovine serum (FBS) from Hangzhou Sijiqing Biological Engineering Materials Co., Ltd. (Hangzhou, China).
-
The wild-type PRV used was vBecker3, generated by transfection of pBecker3 into Vero cells (18). Mutant PRVs (vBeckerZF1 and vBeckerZF2) were constructed by manipulation of pBecker3 and transfection of mammalian cells (see below). All PRV strains were propagated in the PK15 (porcine kidney 15) cell line. Virus titers were determined in duplicate by infectivity end-point assay on PK15 cells in 96-well plates following infection with 100µL/well of 10-fold serially diluted stocks. The mammalian cells were grown in DMEM supplemented with 10% FBS. Viral infections were performed in DMEM supplemented with 2% FBS. Unless otherwise indicated, all media used contained 100U /mL penicillin and 100 µg /mL streptomycin.
-
Constructions of plasmids and BACs are illustrated in Fig. 1. Unless otherwise noted, all PCRs were performed with exTaq hot start polymerase and 2×GC buffer I. The 433 bp of the upstream homology region for allele exchange in E. coli was amplified by PCR from pBecker3 with the primers HRattTn7U-F (5'-GAGCTCCTGCTCCTGGGCTTCCTG-3', with a SacI site underlined) and HRattTn7U-R (5'-GAATTC CCCGTAAGCAAGGCCGTA-3', with an EcoRI site underlined) and cloned into the SacI and EcoRI sites of pEGFP-N1 to generate pZF1, while the 448 bp of the downstream homology region was amplified with the primers HRattTn7D-F (5'-GTCGACGTGCGATC CACGCCCAGC-3', with Sal I site underlined) and HRattTn7D-R (5'-
$\underline {{\rm{GGATCC}}} \underline{\underline {{\rm{ATGCAT}}}} {\rm{GCGCCCAAAGATCTGCCTG}}$ -3', with BamH I site underlined and NsiI site double underlined) and cloned into the Sal I and BamH I sites of pZF1 to generate pZF2. PCR parameters for amplification of both homology regions were as follows: 96℃ for 2 min to denature, followed by 30 cycles of 94℃ for 30 s, 70℃ for 30s, with a final 10-min extension at 72℃. The 712 bp lacZα-mini-attTn7 region was released by Bbs I digestion of pZFBΔtk (unpublished) which contains an 8.6 kb Bsu36I fragment from pHZB10 (19), generating an EcoRI compatible end and a SalI site. Plasmids pZF3 was produced by inserting the lacZα-mini-attTn7 region into the EcoRI and SalI sites of pZF2. To construct the transfer vector for allele exchange in E. coli, a shuttle plasmid pGS284 (17) was used, which includes the π-protein-dependent R6K origin of replication (7), the RP4oriT origin for conjugative transfer (16), and the negative selection marker sacB (11). First, pZF3 was digested by SacI and dephosphorized, and pGS284 was digested by SacI. Then the two linearized plasmids were ligated and the product was transformed into DH5α to select for ampicillin (Amp) resistance. Since pGS284 was unable to grow in a strain that lacks π, only the Amp resistant strains containing the hybrid plasmid pZF4 grew. Plasmid pZF4 was digested by NsiI to excise the origin of replication of pEGFP-N1 and self-ligated to generate the transfer vector pZF5. Shuttle plasmid pFBCMV-GFP, a derivative of pFastBac Dual (Invitrogen), was generated by replacing the P10 and PH promoters with a PCMV promoter and a GFP gene cloned under the promoter.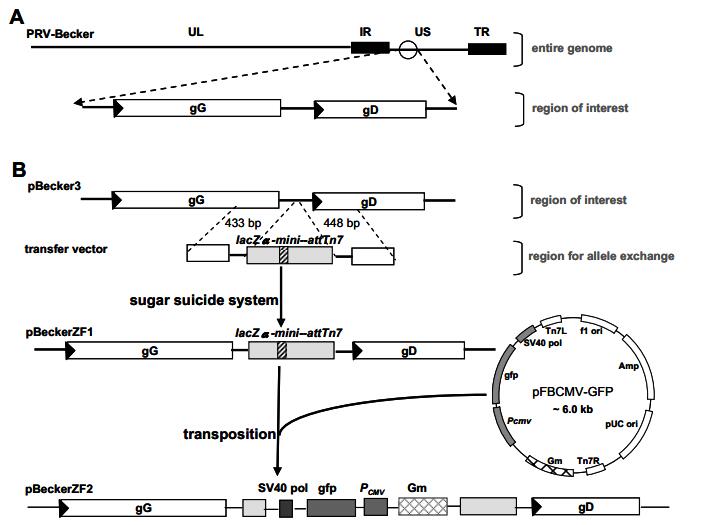
Figure 1. Insertion of lacZα-mini-attTn7 region into pBecker3 and construction of recombinant bacmids by Tn7-mediated transposition are illustrated. A, Schematic diagram of the PRV-Becker genome depicting the unique long (UL), unique short (US), internal repeats (IR), and terminal repeats (TR) regions. A portion is expanded to show the gG and gD genes. For clarity, only this region of all subsequent viral bacmids is illustrated. B, Construction of pBeckerZF1 and pBeckerZF2. Viral genes are represented as rectangles with an arrowhead denoting the transcription direction. lacZα is represented as light gray boxes and attTn7 is represented as hatched boxes.
The pBeckerZF1 BAC was generated by the sugar suicide allele exchange system in E. coli as described previously (17). Briefly, conjugal transfer of the allele exchange vector occurred following cross-streaking of GS500 containing pBecker3 and S17λpir (7) containing pZF5 on a Luria broth (LB) plate without antibiotics and incubated overnight at 30℃. Each intersection from the crossed streaks was inoculated into 5 mL of LB plus 25 µg/mL Amp and 25 µg/mL chloramphenicol (Cm) and incubated overnight at 37℃ with rotating to select for recombinants. To select for deletion of the allele exchange vector from pBecker3, 1 µL of overnight culture was inoculated into 5 mL of LB-Cm and incubated overnight at 37℃ with rotation. The culture was serially diluted (10-3, 10-4, and 10-5), and 50 µL of each dilution was plated onto LB plates containing 5% sucrose, 25 µg/ml Cm, 40 µg/ml isopropyl-β-D-thiogalactopyranoside (IPTG) and 200 µg/mL 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal). A single blue colony was transferred into 5 mL of LB-Cm and incubated overnight at 37℃. Miniprep DNA from 1 mL overnight culture was diluted to low concentration (~ 100ng/mL). The diluted BAC was transformed into E. coli strain DH10B (Invitrogen Co., Carlsbad, USA), precluding co-transformation of more than one copy BAC DNA into a single cell. After one hour incubation at 37℃, transformed cells were spread on LB plates containing Cm, IPTG and X-gal. Blue colonies were further screened for Amp sensitivity then by PCR and sequencing (described below).
The pBeckerZF2 BAC was generated by Tn7-mediated transposition as described in the instruction manual of the Bac-to-BacTM system (Invitrogen Co., Carlsbad, USA). White colonies were picked and further screened by PCR (described below).
-
Virus DNA was obtained from infected PK15 cells as described previously (14). The PCR reactions were set up containing 1×GC buffer I, 200 µmol/L dATP, 200 µmol/L dCTP, 200 µmol/L dGTP, 200 µmol/L dTTP, 0.5 µmol/L concentration of forward primer, 0.5 µmol/L concentration reverse primer and 1U of exTaq hot start polymerase with a total volume of 25µL. For analysis of pBeckerZF1 and vBeckerZF1, two primers flanking the homology regions pBeattTn7-F (5'-GCCCTCTGACATCTTCGTGACCC-3') and pBeattTn7-R (5'-CGAACTTGTACGTGCGGTG CTG-3') were designed and cycling parameters were as follows: 96℃ for 2 min to denature, followed by 30 cycles of 94℃ for 30 s, 70℃ for 1min 50s, with a final 10-min extension at 72℃. For analysis of pBeckerZF2, M13 forward primer (5'-TGTAAAACG ACGGCCAGT-3') and reverse primer (5'-TCACAC AGGAAACAGCTAT GAC -3') were used with the parameters as follows: 96℃ for 2 min to denature, followed by 30 cycles of 94℃ for 30 s, 55℃ for 30 s, 72℃ for 3 min 45s, with a final 10-min extension at 72℃. The PCR products were detected by ethidium bromide staining after separation on 0.8 to 1.0% agarose gels. The PCR product, using pBeattTn7-F and pBeattTn7-R for test of pBeckerZF1, was cloned into pGEM-T Easy and further screened by sequencing.
-
Minipreps of BAC DNA were prepared according to methods developed for the isolation of large plasmids (defined in the Instruction Manual of the Bac-to-BacTM system/Life Technologies). BAC DNA isolated from 1mL of overnight cultures was used for transfection of 40-60% confluent PK15 cells in a 35-mm disk. Transfection was performed with LipofectinTM reagent according to the manufacturer's instructions.
Plasmids, strains, and reagents
Virus and cells
Construction of plasmids and BACs
PCR analysis of recombinant BACs
BAC DNA preparation and reconstitution of infectious viruses
-
Site-specific transposition by transposon Tn7 has been developed to rapidly introduce genes or cis elements into viral bacmids and has proved to be a powerful tool in studies on herpesvirus genetics (2). In this study, to test whether this technology works well in PRV, a recombinant BAC pBeckerZF1 including lacZα-mini-attTn7 was constructed using the E. coli recombination system, which was inserted into the intergenic sequence between the putative polyadenylation signal of the gG gene and the putative TATA signal of the gD gene (3) without loss of any viral genes (Fig. 1 and Fig. 3).

Figure 3. Sequence analysis of lacZ-mini-attTn7 insertion into pBekcer3. lacZ-mini-attTn7 region is in bold and the attTn7 is framed. Homology regions for allele exchange are underlined with polyadenylation signal for gG and TATA signal for gD double underlined.
Insertion of lacZα-mini-attTn7 into the target site in pBecker3 is shown in Fig. 2, evidenced by the different sizes of PCR products with pBeckerZF1 or pBecker3 as template. For PCR analysis of pBeckerZF1, two primers flanking the homologous regions for allele exchange in E. coli were designed to exclude the possibility of contamination by transfer vector pZF5. The size of PCR product with the template pBeckerZF1 is about 710 bp larger than that with pBecker3, which is corresponds exactly with the size of lacZ-mini-attTn7 region (Fig. 2). Further sequencing of the PCR product with the template pBeckerZF1 established that the lacZα-mini-attTn7 region was inserted 44bp downstream from the putative polyadenylation signal of gG and 23bp upstream from the putative TATA signal of gD (Fig. 3.).
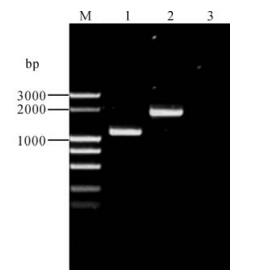
Figure 2. PCR analysis of pBeckerZF1. Two primers flanking the homology regions were used. M, DL3000 DNA marker; 1, pBecker3 as template; 2, pBeckerZF1 as template; 3, Double distilled H2O (ddH2O) as template.
-
To test whether viruses vBeckerZF1 generated from the pBeckerZF1 transfection included the lacZα-mini-attTn7 region, PCR was performed with the same primers used for establishing pBeckZF1 was constructed correctly (see above). The size of PCR product with the vBeckerZF1 genome DNA as the template is about 710 bp larger than that obtained with vBecker3 (data not shown). Therefore, transfection of pBeckerZF1 resulted in the production of virus containing the lacZα-mini-attTn7 region, vBeckerZF1, without the need for further plaque purification.
To examine the growth properties of vBeckerZF1, PK 15 cells were transfected in parallell, with pBeckerZF1 or pBecker3, respectively. Viral titers of the resulted vBeckerZF1 were typically in the order of 108 to 109 TCID50/mL, which is comparable to that obtained for vBecker3. The CPE observed for vBeckerZF1 was also indistinguishable from that observed for vBecker3. Together, these data indicated that the insertion of lacZα-mini-attTn7 in the intergenic region between the gG and gD genes did not visibly affect the viral growth at recorded levels of viral titers and CPE.
-
Three components are necessary for Tn7-mediated transposition: an attachment site, attTn7, a shuttle plasmid (pFastBac Dual or its derivatives) including both the right and left arms of Tn7, and a third plasmid (pMON7124) encoding the necessary trans-acting factors required in transposition (4). In this study the lacZα-mini-attTn7 region containing an attTn7 site in lacZα gene without disrupting its function was used (4). To evaluate the utility of Tn7-mediated transposition for rapid construction of the recombinant virus, a derivative shuttle vector pFastBacCMV-G containing the GFP gene under PCMV was transformed into DH10B harboring both pBeckerZF1 and pMON7124. Transformants were selected on a plate containing Cm, X-Gal and IPTG. White colonies were further screened by PCR with the two primers flanking the attTn7 site. Integration of mini-Tn7 is shown in Fig. 4. The PCR product with pBeckerZF2 as a template was 3.5 kb (lane 2) in length as expected, while that with pBeckerZF1 carrying the intact attTn7 as template was about 300 bp in length (lane 3). Transfection of pBeckerZF2 resulted in production of infectious recombinant virus vBckerZF2 with GFP expression (Fig. 5).
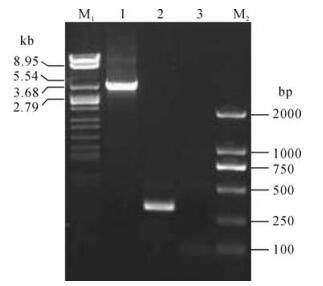
Figure 4. PCR analysis of pBeckerZF2. M13 primers flanking attTn7 were used. M1, λDNA/EcoR I+BamH I+Hind Ⅲ; 1, pBeckerZF2 as template; 2, pBeckerZF1 as template. 3, ddH2O as template; M2, DL2000 DNA marker.

Figure 5. Recovery of infectious vBeckerZF2 by transfection. Pictures were taken 48 hours post transfection. Recovery of vBeckerZF2 was monitored by CPE and GFP expression. A: PK15 cells transfected with pBeckerZF2 exposed to ultraviolet light; B: PK15 cells transfected with pBeckerZF2 exposed to visible light; C: Normal PK15 cells exposed to ultraviolet light; D: Normal PK15 Cells exposed to visible light.
To test whether the mini-Tn7 insertion impaired viral growth, a strategy for examining the growth above). Viral titers and CPE developed by vBeckerZF2 were indistinguishable from that developed by vBecker3. So insertion of at least 3.5 kb mini-Tn7 region into the attTn7 site in vBeckerZF1 did not visibly affect the viral growth at levels of viral titers and CPE.
-
For Tn7-mediated transposition to be useful in herpesvirus genetics, the insertions must be stable in the recombinant virus resulting from transfection. In this study, the stability of mini-Tn7 was tested in cell culture. vBeckerZF2 harvested from transfection of pBeckZF2 was serial passaged in PK15 cells for five rounds at a low motility of infection (MOI) (~0.01 TCID50/cell). The virus harvested was applied to a plaque assay for GFP expression. Of 200 plaques counted, 100% expressed the GFP. This indicates that a 3.5 kb mini-Tn7 insertion by Tn7-mesitad transposition in vBeckerZF2 is stable in cell culture.
Construction of the pBeckerZF1 BAC
Transfection of pBeckerZF1 and characterization of vBeckerZF1 in cell culture
Tn7-mediated transposition for introduction of sequences into PRV bacmid
Stability of mini-Tn7 insertions in vBeckerZF1
-
Members of the Herpesviridae family are characterized with large (130~230kb) double stranded DNA genomes capable of encoding as many as 200 potential genes (12). The genetic analysis of these complex viruses has been a challenge to herpesvirologists. Successful techniques for gene-ration of recombinant herpsviruses have been developed by homologous recombination between the viral genomes and the plasmids containing the desired mutations in infected cells (8, 10). However, these techniques are limited by the fact that wild-type virus is invariably present during the selection process, and, therefore, extensive purification is required to isolate the recombinant virus (5). Mutations which confer a growth disadv-antage to the recombinant virus may make purifi-cation very problematic (5). BAC technology has undoubtedly opened new avenues for mutagenesis of these large virus genomes and enabled the systematic mining of the information stored in virus genomes. The resultant mutant bacmids are clonal and free of contaminating wild type sequences, facilitating rapid production of pure recombinant viruses (2). While this represents a significant advance, mutant construction of these large size bacmids is still time consuming especially when detailed mutagenesis are needed for structure-function studies of viral proteins.
To address this problem, Gabriele Hahn et al adapted a technology employing Tn7-mediated site-specific transposition as a rapid and highly reliable method to create mutants of cytomegalovirus (2). In the present study, we engineered the lacZα-mini-attTn7 region into PRV BAC by homologous recombination in E. coli, and recombi-nant viruses could then be created by transposition of a mini-Tn7 region containing the desired DNA sequences into the attTn7 site. This technology should greatly facilitate detailed mutagenesis of PRV since most of the effort of recombinant virus production will now be located at the point of plasmid construction of the desired mutations.
Since our primary interest lies in functional genomics of PRV, the emphasis of our design was to maintain every gene and element of the parental virus. Thus the lacZ-mini-attTn7 region should be positioned in the viral genome, so as not to interfere with expression of viral genes or function of cis elements. This is not necessarily a straightforward task since the herpesvirus genomes are very compact with few regions that are not transcribed (13). After consideration we therefore chose to place the lacZα-mini-attTn7 region in a relatively large intergenic space between the gG and the gD genes (3). After transfection, the recombinant virus vBeckerZF1 (carrying the lacZα-mini-attTn7 region) and the vBeckerZF2 virus (carrying the 3.5 kb mini-Tn7 insertion region) were recovered and their growth properties were found to be indistinguishable from that of the wild-type virus vBecker3 at the examined levels of viral titers and CPE. Nevertheless, further studies are required to determine the growth properties of vBeckerZF1 and vBeckerZF2 in tissue cultures and their virulence in animals.
Stability of the insertions in recombinant viruses generated by BAC technology should be taken into consideration. In studies reported by Smith and Enquist, F-plasmid sequences inserted in the gG ORF of PRV were unstable following passage, however, the lacZ insertion at the same locus or the F-plasmid insertion at another locus showed no signs of instability (17, 18). More information is needed to understand these differences. We tested the stability of mini-Tn7 insertions in the intergenic region between the gG and gD genes and results showed that insertion of a 3.5 kb mini-Tn7 into vBecker1 by transposition was stable in cell culture. Future work is needed to make clear whether larger insertions or insertions of different sequences into vBeckerZF1 are still stable.
The size of insertions should also be taken into consideration. It is yet unclear how many heterologous genes may be packaged into the BACderived viruses before size constraints become problematic. Although HSV can package up to 30 kb in excess of its genome, the packaging constraints of other herpesviruses are yet unknown. Initial studies with MCMV BACs did report stability problems once more than 5 kb of extra DNA was inserted into the viral genome (5). In this study, to minimize the size of insertion, the lacZα-mini-attTn7 region was inserted alone but not together with an antibiotic resistance gene (which is standard for homologous recombination in E. coli). Although more complicated procedures were needed to get the clonal recombinant bacmid, this did permit a capacity for accommodating larger insertions.
In summary, a technology employing Tn7-mediated site-specific transposition as a rapid method to introduce novel sequences into bacmidcloned PRV genome was developed and which allowed recombinant viruses to be rapidly and reliably created. This technology should accelerate greatly the pace at which recombinant viruses can be created and, thus, facilitate the use of recombinant viruses for detailed mutagenic studies.

 DownLoad:
DownLoad: