









-
Genetic diseases can occur in various ways with complex causes, but conventional therapies are very limited or non-existent in many instances. Gene therapy approaches attempting to subvert, correct or prevent the pathogenic mechanisms of specific genetic disease by the introduction of corrective genetic information into appropriate target living cells represent a good alternative. It has been over 30 years since the first proposal of using gene therapy as a therapeutic method for the treatment of genetically determined inherited diseases (21). The idea of treating human diseases by introducing functional genes into living cells to achieve therapeutic purpose has now become a clinical reality (3). The first successful attempt of using molecular-based therapeutic techniques for the treatment of human recessive hereditary deficiency was documented in 2000 when Cavazzana-Calvo and his co-workers demonstrated they were able to overcome severe combined immunodeficiency (SCID-X1), an X-linked inherited disorder in children, through retrovirusmediated transfer of complementary DNA containing a gamma c and ex vivo infection of CD34+ cells (8). Successful completion of this clinical gene therapy showed great promise for the use of gene therapy to provide full correction of genetic disease phenotype and associated clinical benefits. Today, human gene therapy represents a promising new form of medicine which is under active development. The potential application of gene transfer technology has now been extended to the treatment of a variety of diseases including infectious diseases, AIDS, neurological disorders and cancer (7, 20, 27, 35).
Successful gene therapy approaches are largely dependent on the development of safe and efficient gene-delivery systems to transport therapeutic genes into the target destination. The vectors, or gene delivery systems, play a crucial role as effective tools for genetic modification of the majority of somatic cells in vitro and in vivo in the development of human gene therapy protocols (12, 25, 42). Vectors derived from a variety of viruses including retroviruses, adenoviruses, adeno-associated viruses, baculoviruses and herpesviruses, are currently being developed and evaluated for their potential use as gene transfer vehicles (27). Among these viral vectors, genetransfer vectors based on lentiviruses, particularly HIV-1-derived vectors, have been widely used for gene therapy applications since their initial construction in 1991 (36) and the interest in using such systems in applied settings will continue to grow (18, 31, 46). Present studies have shown that HIV-1-based vectors are attractive gene delivery tools due to their relatively large coding capacity, efficient gene transfer, ability to establish long-lasting transgene expression, ability to integrate into genomes of nondividing cells and to inhibit wild type HIV-1 replication in the absence of any anti-HIV-1 insert (2, 4, 5, 11, 42). Because of the potential for possible clinical gene transfer applications in the future, lentiviral vectors mediated gene transfer have been tested for their ability to infect various types of cells in vitro, in vivo and ex vivo, including hepatocytes (19, 41), hematopoietic cells (1, 14), stem cells (30, 40), monocyte-derived dendritic cells (22), lymphocytes (13, 47), monocytes/macrophages (29, 34, 38), and neurons (33, 44). All these cells are important targets in human gene therapy (4, 25, 35).
To further explore the use of a lentiviral vector as a potential gene transfer tool in human gene therapy, it would be necessary and important to develop an in vitro transfection protocol for consistent production of high yields of vector virus. The objectives of this study are directed towards optimization of in vitro protocols for DLV vector production and vector mediated gene transduction. We have established a method for preparation of high-titer vector and vector concentration through a simple one-step ultracentrifugation. Constructed HIV-1-based vectors pseudotyped with a VSV-G envelope protein are highly infectious to human T-cell lines and cells derived from a variety of mammalian species. DLV-mediated gene delivery into human T-cells facilitate a stable and long-term transgene expression and transductant cells become refractory to the infection by wild type HIV-1, suggesting that the transduced cells are protected from HIV-1 infection. We also demonstrated that these vectors are likely to be safe to use since we detected no generation of replication-competent virus (RCV) through vector recombination in transduced cells.
HTML
-
All three HIV-1-based defective vectors, DHIV (DHIV-Rev+), DHIV-Rev-and DHIV-CTE, encoding selected cis-acting elements, express a reporter gene, green fluorescent protein (GFP), under the control of HIV-1 LTR. The vector, DHIV-Rev+, has deletions that affect structural (gag/pol and env) and accessory genes (vif, vpr, and nef), but contains all cis-acting elements required for vector packaging and transduction (43). The vector, DHIV-Rev-, has a deletion in the second coding exon of Rev in DHIV-Rev+. The rev-independent vector, DHIV-CTE, was produced by replacing the RRE in DHIV-Rev- with the constitutive transport element (CTE) from Moson-Pfizer monkey virus (26, 29). All plasmids were prepared using Qiagen plasmid-Prep kits (Qiagen). DNA yield was determined by measuring the concentration of DNA in the elute by recording the absorbance at 260 nm in a Beckman DU-640 spectrophotometer (Beckman).
-
Human T-cell line, CEM (human T4-lymphoblastoid cell line, obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH), COS-7 (Africa green monkey kidney cell) and Sup-T1 (Non-Hodgkin's T-cell lymphoma cells) were propagated in RPMI-1640 medium (Sigma), while 293T (Human embryonic kidney cell), NIH3T3 (Mouse embryo cell), MT-2 (Human T-cell leukemia cell) and PA 317 (Mouse embryo hybridoma cell) cells were maintained in Debecco's Modified Eagle's Medium (Sigma) and Vero (Monkey kidney cell), HeLa (Human cervix epithelial adenocarcinoma cell), WI38 (Human lung fibroblast cell) and HTB-14 (Human glioblastoma cell)cells were maintained in Minimum Essential Medium Eagle (Sigma). All these cell cultures were incubated at 37℃ with 5% CO2 in proper media supplemented with 10% heat-inactivated fetal bovine serum (FBS) (HyClone), 100U/mL penicillin, 100ug/mL streptomycin, and 0.292mg/mL L-glutamine (Sigma). Cells were observed and documented using a phase-contrast inverted microscope (Olympus IX70).
-
HIV-1-based vectors pseudotyped with VSV-G were produced by transiently transfecting human embryonic kidney 293T cells with a packaging construct, a VSV-G envelope construct and a transfer construct containing GFP gene (43). Twenty four hours before transfection, 293T packaging cells at exponential growth phase were subcultured in T-75 flasks (Corning) at a density of 6×106 cells/flask and incubated at 37℃ with 5% CO2. The growth medium was replaced with fresh medium 2-3 h prior to the transfection. DNA mixture containing 6.25 µg packaging plasmid, 6.25 µg transfer plasmid and 2.50 µg VSV-G plasmid was prepared per TC-75 cm2 flask and added to the cell monolayer drop-by-drop. Chloroquine solution was added immediately after to yield a final concentration of 25 µM/mL. Eight hours following the transfection, the growth medium was replaced with 8 mL/flask of fresh DMEM supplemented with 2% FBS. Vector production at post transfection days 1, 2, 3 was titered in CEM cells using 10-fold serial dilution method. Vector titer was determined by visually counting the GFP positive cells of the endpoint dilution using fluorescence microscopy (23, 28).
-
Preparation of large-scale production of HIV-1 based vectors was conducted in TC-150 cm2 flasks (Corning) using the same protocol. 293T packaging cells were seeded at 1.2 -1.5×107 cells/flask in 25 mL DMEM medium 24 h prior to transfection and 6-10 flasks were used each time. Transfection was conducted using double concentrated DNA mixture and vector produced from different batches were stored at –80℃. To concentrate DLV, vectors stored at –80℃ were thawed and pooled. Following a low speed centrifugation (1 800×g) for 30 min at 4℃, recovered supernatant was filtered through a 0.45 µm filter unit to remove cellular debris (Nalgene). Filtrate containing the vector was ultra-centrifuged at 113 000 ×g for 2.5 h at 4℃ using a Beckman SW28 rotor. Vector pellet in each tube was resuspended in 0.2mL RPMI-1640 medium containing no FBS or antibiotics. Vector aliquots (0.1-0.2 mL/tube) were stored at –80℃ for future titration and transduction.
-
The transduction efficiency of HIV-1-based defective vectors for human T-cells (CEM) was tested using different vector multiplicities of infection (MOI) of 1, 5, 20 and 50. CEM cells at their exponential growth phase were harvested and counted. For DLV transduction, 1.0×105 cells were pelleted in a 1.5-mL sterile tube and resuspended with 0.5 mL of diluted DLV stocks. Following 1 hr vector adsorption at the presence of 8 µg/mL polybrene at 37 ℃ and gently tapping the tube every 15 min, the cells were washed once, and then resuspended with 1.0 mL medium supplemented with 5% heat inactivated FBS and seeded into 2 wells of a 24-well plate (0.5 mL/well). Following seventy-two hours incubation, GFP positive cells were visually counted under a fluorescence microscopy and the transduction efficiency were determined by recording the percentage of the GFP+ cells within a transduced cell population.
-
11 cell lines commonly used for gene transduction including CEM, MT2, Sup-T1, COS-7, PA317, 293T, NIH3T3, HeLa, Vero, WI38 and HTB-14 were tested and compared for their sensitivity to DLV infection in vitro. All the cells were harvested at their exponential growth phase and the concentration was adjusted to 2.0×105 cells/mL. A DLV stock was 10-fold diluted (10-1 to 10-6) with mediumcontaining 8μg/mL Polybrene. DLV transduction was conducted by resuspending different cell lines with 1.0 mL of the same DLV dilution. Following 1 h adsorption, infected cell suspension was inoculated into a 96-well plate, with 0.1 mL/well cells (2.0 × 104 cells/well), and 4 wells/dilution/cell line. The 96-well plates were covered with sensitive films and incubated at 37℃ with 5% CO2. At post infection day 3, GFP+ cells were visually counted under a fluorescence microscopy and the sensitivity of these cells to DLV infection was determined by examining the calculated vector titer (averaged of 4 well at endpoint dilution).
-
CEM cells transduced with three HIV-1-based defective vectors were cloned using the limited dilution method. In brief, transduced cells were diluted with the conditioned RPMI growth medium to the concentration of 10 cells/mL, and then inoculated at 0.1 mL/well into a 96-well plate. Following a period of 4-day cultivation at 37℃, GFP positive clones derived from a single cell were identified, transferred into a 12-well plate after 7-10 days and then transferred into TC-25 cm2 flasks (Corning). The long-term expression of GFP gene was determined by microscopical examination of transduced cells at different passage times (1 week to 1.5 year) and/or by PCR and RT-PCR detection.
-
To evaluate if replication-competent virus is generated in transduced cells through vector recombination, two independent methods were used: 1) p24 assay to measure virus production in transduced cells. In brief, medium specimens were collected from DLV-transduced CEM cells at different passages (10~40 passages) and stored at –80℃. RCV generation in the medium was determined by measuring viral p24 antigen production (Coulter, Beckman). 2) Infectivity assay to measure infectious RCV. In this assay, collected medium specimens were filtrated through 0.22 µM filters to remove cellular debris and dead cells. Recovered filtrates were diluted with equal volume of fresh growth medium RPMI-1640 (for CEM and Sup-T1 cells) or DMEM (for MT-2 cells) and then used to infect these indicator cells for a period of at least 1 month. The emergence of GFP positive cells was checked every other day under a fluorescence microscopy.
-
Three cell populations (including a DLV-transduced GFP+ clone, normal CEM, a mixed cell population containing 1/2 GFP+ clone and 1/2 normal CEM cells) were infected with a same HIV-1 stock (MOI = 0.1) in the presence of 8 µg/mL polybrene. Following 1 h adsorption at 37℃, cells were washed twice with DPBS and seeded into TC-25 cm2 flasks. Syncytial formation induced by HIV-1 infection was examined daily and viral replication was assayed by measuring p24 viral antigen production (ELISA kit from Coulter). Cell viability was determined by trypan blue staining and GFP+ cells were visually counted using fluorescence microscopy.
-
Measurement of DLV mobilization was conducted in two ways. 1) Direct measurement of the increase of GFP+ cells in a transduced CEM population. DLVtransduced CEM cells and a GFP+ clone were mixed with normal CEM cells to generate two sets of CEM populations containing 10%, 30% and 50% GFP+ cells. These cells together with normal CEM and GFP+ clone were super-infected with wild-type HIV-1 -IIIB at a MOI of 0.1. The vector mobilization was determined by the increase of the number of counted GFP+ cells within the cell population. 2) Infection of normal CEM cells with conditioned medium. Culture supernatant from the above super-infected groups was collected and centrifuged at 1 800 g, 4℃ for 30 min, then filtered through a 0.22 µm filter (Costar). Conditioned media were prepared by mixing with these cell-free culture supernatants with fresh medium at the ratio of 1:1. Normal CEM cells were cultured with this conditioned media and the emergence of GFP positive cells were checked by fluorescent microscopy.
Plasmids
Cell cultures
Vector production
Vector concentration
Transduction efficiency of DLV in CEM cells
Transduction with DLV in different cell lines
GFP+ cell Cloning and long-term expression of GFP
Assays for replication-competent virus (RCV)
Inhibition of wild type HIV-1 replication
Vector mobilization
-
The DLV pseudotyped with the VSV-G envelope protein used in this study was generated in 293T cells according to the well-established calcium phosphate co-precipitation method. Transfection was evidenced by the presence of large percentage of GFP+cells (>70%) at day 1 post-transfection which extended to the whole cell population by day 3. This was accompanied by rapid increase of vector yield from 5.6×106 IU/mL at day 1 to 10.7×106 IU/mL at day 3 (Fig. 1A). To define the vector production by time, vector generated from each post-transfection day was separately quantified. Although many vectors were assembled shortly after transfection (5-7.5 × 106 IU/mL at day 1), the majority of vectors were produced at day 2 (>10×106 IU/mL) as shown in Fig. 1B. Vector production dropped to less than 6×106 IU/mL at day 3. Because of the cellular fusion and detachment of large percent of transfected cells, vector production was dramatically reduced at transfection day 4 and subsequent days (data not shown).
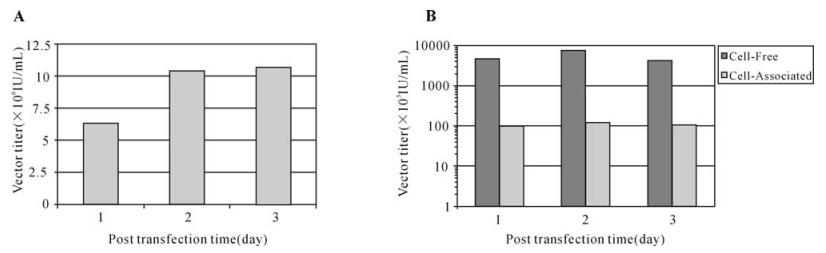
Figure 1. DLV (DHIV-Rev-) production in 293T packaging cell line DLV vector was produced under the same transfection protocol and titration of vector by limited dilution infectivity assay using CEM cells. A: Accumulative DLV production measured at selected post transfection time. B: Comparative analysis of vector produced in cell-free medium supernatant and associated within the 293T packaging cells at different post transfection times. The result of each test represented an average of three experiments.
To define the localization of DLV, we comparatively analyzed daily production of DLV released into cell culture medium (cell-free vector) and vectors associated with the packaging cells (cell-associated vector). The majority of assembled DLV (>97.5%) was released in the cell-free form in the cell culture medium while only a small portion of vector ( < 2.5%) was associated with the packaging cells (Fig. 1B). This led to the exclusion of the cellular pellet to be used for vector isolation and concentration.
To generate high-titer vector stocks, we have established a one-step ultracentrifugation protocol to concentrate vector produced in large-scale or from different batches of viral transfection. As shown in Table 1, the vector virus was effectively concentrated using this method and concentrated vector yielded a final titer up to 9.83±2.25×108 IU/mL. This vector concentration method is relative simple, rapid, easily reproducible, and it allowed a high recovery rate of infectious vector (90%) (Table 1).

Table 1. DLV production in 293T cells and concentration by ultracentrifugation
-
All three DLV vectors were capable of infecting CEM cells and they shared the same pattern of transduction efficiency, which is largely related to the concentration of DLV stocks.
As shown in Fig. 2, approximately 30% cells were successfully transduced when a low-titer of DLV stock was used (MOI = 1.0). This transduction efficiency increased to about 60% at a MOI of 5 and reached over 80% at a MOI of 20, indicating that the transduction efficiency is directly correlated with DLV titer. More than 90% of CEM cells became GFP+ when a more concentrated DLV preparation (MOI = 50) was used for the transduction. In addition, human glioblastoma cells (HTB-14) available in this laboratory were also tested for their susceptibility for DLV transduction. We demonstrated that DLV were highly infectious to HTB-14 cells with a transduction efficiency of 50% at MOI of 1.0 and over 90% at MOI 20 (data not shown). This finding may suggest DLV have the ability to transduce human microglial cells, which are very important target cells of HIV-1 infection in the human central nervous system.
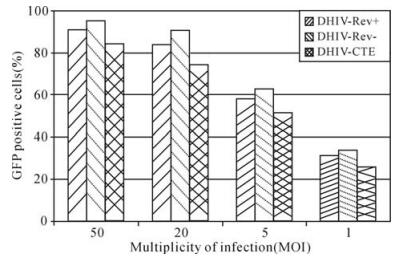
Figure 2. Transduction efficiency with DLV in human Tlymphocyte cell line CEM. Each transduction was conducted with the use of 1.0×105 cells and transduction efficiency was determined at post infection day 3. The result of each test represented an average of two experiments.
-
Infectivity of these DLV to other cell lines were determined and compared by the titration of the same DLV preparation in ten different cells derived from human and other mammalian species. All these cells appeared to be susceptible to DLV infection. However, efficiency of DLV-mediated gene transduction of these cells differed remarkably (>10 000 times). As shown in Fig. 3, human lymphoid cells (CEM, MT-2, SupT1 and HTB-14) were the most sensitive to DLV infection while HeLa, 293T and WI38 cells were comparatively less sensitive, and murine-derived NIH3T3 and PA317 cells were the least sensitive. The ability to infect a wide spectrum of cell types suggests the potential use of these DLV pseudotyped with VSV-G in gene transfer for human and other mammalian species.
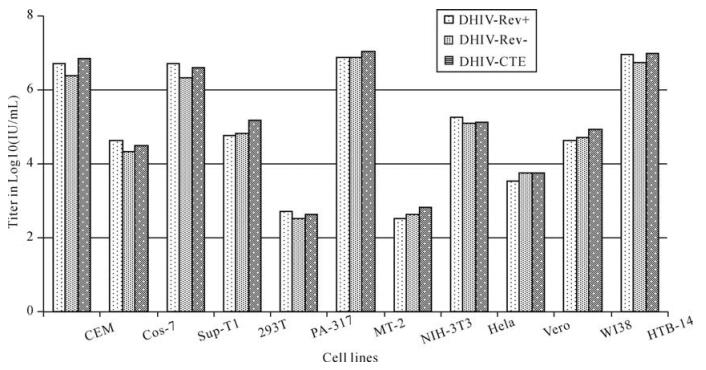
Figure 3. Transduction of different cell lines with DLV. Cells were harvested at their exponential growth phase and adjusted to a concentration of 2.0×105 cells/mL. 1 mL of this cell preparation was pelleted down an Eppendorf tube and for each vector 6 tubes of this cell pellet were prepared. Master preparation of DLV was 10-fold serially diluted (10-1 to 10-6) with serum-free medium containing 8ug/mL Polybrene. Infection of cells with DLV was conducted by resuspending the prepared cell pellet with 1.0 mL DLV preparation from 10-1 to 10-6, separately. Following 1 h adsorption, infected cells was inoculated into a 96-well plate at a concentration of 0.1 mL/well for cells (2.0×104 cells/well) and 4 wells/dilution of vector for each cell line. The 96-well plate was covered with sensitive film and incubated at 37℃ with 5% CO2. At day 3 post infection, GFP+ cells were visually counted and documented under either normal or fluorescent light (Olympus IX 70) by using mounted Olympus digital camera and software MagnaFire. The infectivity of DLV to 11 cells to DLV was determined by comparing vector titer (average of GFP positive cells from 4 wells at the endpoint dilution).
-
Cloning culture of DLV-transduced CEM cells were conducted in 96-well plates using the limited dilution method. The GFP positive cell clones derived from individual transduced cells were cultured with conditioned medium prepared from normal CEM cultures. By days 10-14, clonal cultures formed sizable cell colonies, then were transferred to a 12-well plate for 7-10 days before propagated in TC-25 cm2 flasks. Clonal cells were subcultured more than 60 times in vitro in a period of 1.5 years and showed no change in GFP expression in transduced cells (Fig. 4A, fluorescence light; 4B, normal light). This long-term stable transgene expression was also demonstrated by analyzing nucleic acid extracted from transduced CEM cells by RT-PCR (Fig. 4C).
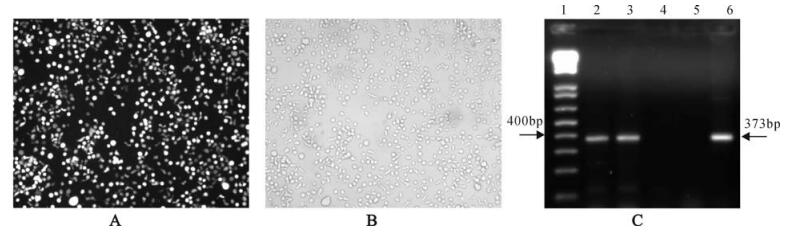
Figure 4. Stable long-term expression of GFP gene in DLV-transduced CEM cells. A: Photomicrograph of GFP+ transductant CEM cells at passage 60 under fluorescence light; B: Photomicrograph of GFP+ transductant CEM cells at passage 60 under normal light; C: RT-PCR detection of transgene (GFP) expression in transductant CEM cells. Lane 1: 1kb plus DNA ladder (Invitrogen); 2: Total RNA from the 9th passage DLV-transduced CEM; 3: Total RNA fron the 48th DLV-transduced CEM; 4: Untransduced CEM; 5: Negative Control (water); 6: Positive control (plasmid DHIV-Rev-). Gel running: 2% agarose, 50V 90min, EB staining, Bio-Rad FX molecular imager scanning. Target fragment size: 373bp.
-
The potential for the generation of RCV through vector recombination during vector production, transduction and long-term culture of transduced cells was investigated using both p24 ELISA and viral infectivity assays. We have detected no RCV in all the samples we tested (data not shown). Initially, we examined the conditioned media collected from both early (8-10 passages) and later (>40 passages) cultures of different clones of DLV-transduced CEM cells and p24 antigen was not detected in these clones over a period of 1 year. The medium samples were then tested for infectious virus by inoculating the media to several human T-lymphocytes including CEM, SupT1 and MT-2 cells. Following in vitro cultivation for at least one month, these cultures showed neither any GFP+cells nor p24 production. Over a dozen of the transduced cultures were analyzed and generation of RCV was not detected (Data not shown). In addition, the negative detection of RCV was not due to the use of lower DLV preparations since present study included the use of 5 different DLV stocks with an average vector titer of more than 109 IU/mL.
-
The lentiviral vector-mediated inhibitory effect on viral replication was demonstrated by challenging transduced CEM with a wild type HIV-1 strain (Fig. 5). When normal CEM was infected with HIV-1, viral replication was detected shortly ( < day 5) and followed by rapid increase of viral p24 production which was evidenced by the massive formation of typical ballooning-shaped cells (syncytia). Viral replication reached a peak by post infection day 15 and then slowed down gradually due to the death of affected cells. When the same HIV-1 stock was inoculated into a transduced (50% GFP+) CEM population, HIV-1 replication was partially inhibited. This limited antiHIV-1 infection was determined by the delayed detection of viral replication, reduced viral production (p24) and limited level of syncytial formation. In comparison, HIV-1 replication was completely inhibited when GFP+ clones were challenged with the same viral stock. Similar results were also obtained when these clones were challenged with a more concentrated HIV-1 dose (MOI = 1.0). Further analysis of these infected cells revealed that GFP+ clones showed a consistent high percentage of cell viability (>92%) during the course of infection. In contrast, HIV-1 infection resulted in significant drop of cell viability to less than 15% for control CEM at postinfection day 21 as compared to 60% for the mixed population (Fig. 6A). The percentage of GFP+ cells gradually increased from the initial 50% to 80% following HIV-1 infection while no change was observed for the uninfected population (Fig. 6B). These data clearly suggest the resistance of GFP+ cells to HIV-1 infection and possibly vector mobilization.
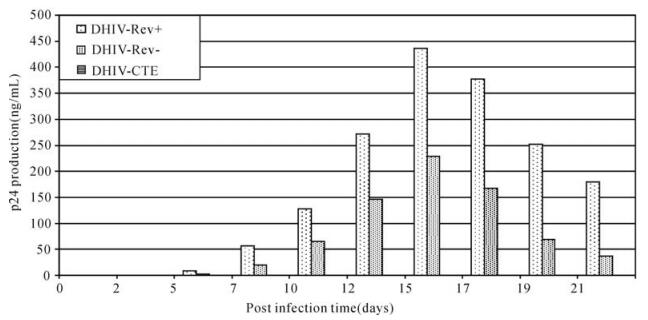
Figure 5. Inhibitory effect of DLV on HIV-1IIIB replication. Viral infection of normal CEM (CEM-N) was monitored and compared with the same infection conducted in both a DLV-transduced CEM clone (GFP+ clone) and a cell population composing of equal number of normal CEM and GFP+ cells. Data show HIV-1 replication was significantly suppressed in the mixed culture and completely inhibited in the GFP+ clone.
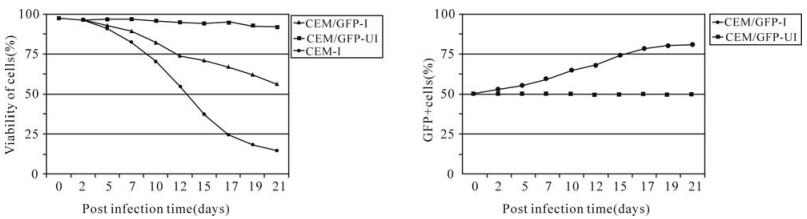
Figure 6. Comparative analysis of cell viability and percentage of a transduced CEM cell population (50% GFP+) infected (I) or Uninfected (UI) with HIV-1IIIB. A: Kinetics of cell viability of a 50% GFP+ CEM cells compared to normal CEM cells. B: Percentage of GFP+ cells gradually increased with HIV-1 infection compared to no change for the uninfected cell population. CEM/GFP-I = HIV-1 infected transducatant CEM (50% GFP+) cells, CEM-GFP-UI = Transductant CEM (50% GFP+) cells without HIV-1 infection, CEM-I = normal CEM cells infected with HIV-1.
To define vector mobilization to untransduced cells, partially transduced CEM cells containing 10%, 30% and 50% GFP+ cells were infected with a low dose of HIV-1 (MOI = 0.01). We observed that the initial percentage of GFP+ cells increased very slowly for the 10% GFP+ population but rapidly increased for the 50% GFP+ group, and reached more than 75% by day 21. Examination of the normal CEM cells cultured with the conditioned media prepared from 50% GFP+ cells infected with HIV-1 revealed the appearance of GFP+ cells (Data not shown). This phenomenon was also observed when Sup-T1 and MT-2 cells were cultured with the cell-free conditioned medium. These data indicated the generation of replication-competent DLV in the HIV-1 infected transductant cultures, which confirmed the mobilzation of DLV.
Vector production and concentration
DLV-mediated transduction efficiency
Transduction of vectors in different cell lines
Cloning of transduced cells and long-term expression of the GFP gene
Generation of replication-competent virus (RCV)
Inhibition of wild-type HIV-1 replication and vector mobilization
-
The application of viral vectors as a gene transfer systems play a crucial role in the development of human gene therapy protocols. Lentiviral vectors based on the HIV-1-genome are emerging as the most attractive and promising vehicles for delivering therapeutic genes into human target cells. These vectors have the ability to efficiently integrate into a wide variety of cells and provide a highly efficient, stable environment for long-term transgene expression. One of major factors affecting DLV-mediated gene transduction of target cells is the production of high titer vector virus. 293T packaging cell line derived from human kidney has been the most widely used vector-producing system for HIV-1-based vector (5, 10, 17, 23, 26, 29, 34, 37). To enhance the production of high-titer of DLV in 293T cells, it is very important to seed cells at the exponential growth phase at the appropriate density. Our data show that seeding 6.5× 106 cells per TC-75 cm2 flask one day prior to the transfection led to the formation of an approximately 70% cell monolayer in the next day. This cell density appeared to be superior to the other two cell seeding densities (50% and 95%) for the production of lentiviral vector.
Vector systems based on lentiviruses has been an important tool for human gene therapy. However, use of these vectors for gene transfer was limited for a number of years initially because of several vectorrelated problems including low vector titers and narrow host ranges. It was reported that the innate envelopes of lentiviruses are much more fragile and attempts to concentrate these vectors through ultracentrifugation would cause the loss of vector infectivity (1, 6). Over time, advances in the understanding of lentiviral vector design and gene transfer have occurred and progress has been made in overcoming the identified limitations. Present studies have shown that vectors based on murine retroviruses and HIV-1 can be pseudotyped with an amphotropic envelope glycoprotein. In particular, the G-protein of vesicular stomatitis virus (VSV-G), a single-stranded RNA virus belonging to the rhabdoviridae family, has been widely used for vector production. Because of much wider spectrum of receptors for VSV-G including ubiquitous anionic phospholipids, the pseudotyped vectors exhibit a much broader host cell range than vectors using the conventional amphotropic env (6, 16, 39). More importantly, because of the physical stability of VSV-G-containing envelopes, it is possible to concentrate pseudotyped vectors by a hundred and thousand fold through ultracentrifugation (1, 6, 10, 29, 45). We have established a protocol for effective concentration of DLV which includes the use of 0.45 µm filters to remove small cellular debris and fragments and the addition of small volume of 50% sucrose which acts as a cushion in the centrifuge tubes to allow the formation of a vector pellet which could be readily resuspended in small volume of buffer. Under these laboratory conditions, we can easily concentrate DLV over 100-fold with the recovery rate as high as 90%. This vector concentration protocol has been routinely performed in this laboratory and the concentrated vector titer is in the range of 0.50-1.25× 109 IU/mL.
Production of high yield vector is crucially important and essential for human gene therapy because high titer of vector stocks promotes high efficiency of gene transfer. This means that a high percentage of transduced target cells can be readily obtained without going through a long-time drugselection step. Furthermore, high efficiency of gene transfer is of great importance in human gene therapy since it allows a shortened in vitro cell manipulation time, which would largely reduce the risk of the biological characteristics and function of target cells. We have shown that the efficiency in transducing human lymphoid cells is directly correlated with the titer of vector virus. As we demonstrated in this study, DLV-mediated transduction efficiency reached over 90% at a MOI of 50, compared to only 30% when target cells were transduced with a lower titer of vector stock (MOI = 1).
There are several major concerns regarding the use of HIV-1 based vector as gene delivery system, including the possible adverse generation of RCV. These concerns have led to the new design and production of self-inactivating lentiviral vectors and vectors containing different viral elements with improved safety (32, 43, 48). We have examined the generation of the RCV from five batches of vector production with an average titer of approximately 109 IU/mL; the results showed no RCV were generated. To date, generation of infectious virus has not been reported from second or third generation lentiviral vectors (24), and it is unlikely that we will observe the presence of RCV in the DLV-transduced CEM cells in the 1.5 years duration of this study. Together with the demonstration of unchanged morphology and growth kinetics of transductant cells, our preliminary data suggest the constructed DLV represents a safe gene transfer system (9, 17). However, further analysis of these vectors would be necessary using other cell culture systems and animal models prior to clinical testing.
Several investigators have recently reported that HIV-1-based defective vectors can efficiently inhibit wild-type HIV-1 replication (2, 4, 5, 11, 42). In particular, this occurs when the vectors do not encode any specifically designed antiviral insert. We have demonstrated stable transduction of various cell types with HIV-1-based vectors. Analysis of transductant CEM cells showed that they are refractory to HIV-1 infection; this was observed by monitoring p24 production, syncytial formation and cell viability counting. Potential mechanisms of this DLV-mediated anti-HIV-1 effect include TAR and RRE decoy effects of the vectors, the competition of vectors for the substrate necessary for reverse transcription and RNA encapsidation (2, 5, 15). Another possible reason for the inhibition of HIV-1 replication involves the formation of defective HIV/vector recombinants caused by the dimerization of HIV-1 and vector RNAs at the dimmer linkage structure (DLS). The defective heterodimers could be packaged into virions and responsible for HIV-1 inhibition by either competing for the packaging or, later, interfering with the strand transfer (34). All these suggest the occurrence of some fatal changes in virus infectivity after the wild-type HIV-1 enters the transduced cells. In addition, demonstration of vector mobilization indicated the successful package of defective vector genome in the newly assembled virus particles transduced cells, either in heterologous HIV/vector recombinant RNA or in homologous vector/vector RNA. Because of the relatively smaller size, it is possible that defective vector genome is somewhat more preferably packaged in encapsulation. Thus the transduced cells could serve as a filter to remove the toxicity of the wild-type HIV-1(5, 26).
Another interesting property of the vectors is their ability to be trafficked by wild-type HIV-1 to untransduced cells (26). These HIV-1-based vectors are engineered to contain cis-acting elements necessary for replication and packaging (i.e., the long terminal repeats and the primer binding site). Therefore, they have the potential to be packaged into the HIV-1 capsid and envelope to form new virus particles with the vector genome. We have employed two methods to determine the vector mobilization and demonstrated the trafficking of DLV into the untransduced cells following a low-dose HIV-1 infection. This property may prove to be extremely useful for the purpose of HIV-1 gene therapy which allows the protection of a large number of target cells by initially transducing a small number of cells.
In conclusion, considering the high transduction efficiency in various cell types, stable transgene expression, no generation of RCV, and anti-HIV activity, these newly constructed HIV-1-based vectors are represent a valuable gene transfer system for human gene therapy. We are currently characterizing these vectors interaction with wild-type virus to understand the molecular mechanisms of their antiHIV-1 effect.

 DownLoad:
DownLoad: