











-
Baculoviridae is a family of large enveloped viruses with double-stranded, covalently-closed, circular DNA genomes (80-180 kb) that infect arthropods exclusively and primarily insects (52). Baculoviruses are characterized by rod-shaped enveloped nucleocapsids and two phenotypes of virions are typically evident during the replication cycle in host insects. The occlusion-derived virion (ODV) is occluded in a crystalline protein matrix during the final stages of virus replication in the nucleus of infected cells, and the budded virion (BV) that is formed as nucleocapsids bud through the plasma membrane of infected cells. Budded virions are responsible for virus spread to tissues throughout the host producing a systemic infection, and ODV are responsible for horizontal transmission of baculovirus from host to host upon ingestion of the virus occlusion body (OB). There are four genera recognized based on ODV-OB morphology, the taxonomic ORDER of susceptible insect hosts and genome sequence phylogenic analysis; these include: Alpha-, Gamma- and Deltabaculovirus (formally known collectively as the genus Nucleopoly-hedrovirus [NPV]) and Betabaculovirus (formally known as the genus Granulovirus) (26). Historically, baculovirus species have been named on the basis of the host from which they were isolated. This is a system of nomenclature that is not without its problems as multiple baculovirus species have been derived from the same host species; for example, Mamestra configurata nucleopolyhedrovirus (MacoNPV) species A and B (33). In addition, the same baculovirus species can be derived from field populations of multiple host insect species; for example, Xestia c-nigrum granulovirus (XecnGV) has been isolated from at least five noctuid species (19). Species designations within Baculoviridae are currently based on host range and restriction endonuclease (REN) profiles of genomic DNA, and increasingly DNA sequence analysis is used to differentiate species (52).
Baculoviruses have long been recognized as potential biological control agents for insect pests based on their causal association with spectacular epizootics, particularly among lepidopteran and sawfly forest pests. Interest in baculoviruses as potential biopesticides naturally led to efforts to screen insect populations for new and potentially more efficacious isolates or strains. With the application of new molecular tools such as restriction endonucleases (REN) and targeted polymerase chain reaction (PCR) technology to characterize baculoviruses it became evident that multiple genotypes occur among baculovirus populations in field isolates derived from different geographic or temporal populations of a single host insect species. Genotypically distinct strains from these populations have been derived from field isolates either by plaque purification in permissive cell lines (28) or upon in vivo cloning (41, 49). Not only are genetic variants/strains common in pooled samples of host insect larvae from a geographic population of insects but substantial numbers of genetic variants of a baculovirus species can occur in single infected individuals (9). Genetic variation in field populations has typically been detected by means of restriction fragment length polymorphisms (RFLP) and purified strains from these populations often have been demonstrated to have differences in infectivity or virulence both in vivo and in vitro. Advancements in DNA sequencing technology are beginning to allow the complete sequence analysis of multiple strains of a baculovirus species and therefore detailed information on genetic variation including patterns of variation are beginning to emerge. In this short review specific examples of the characterization of baculoviruses field isolates with respect to infectivity and pathogenicity, as well as, genetic variation will be used to demonstrate some general trends. Where examples exist, the comparisons of complete genome sequence of different isolates of a baculovirus species will be reviewed in order to examine common regions of genetic variability, and potential mechanisms that maintain genetic diversity in baculovirus populations will be discussed. Finally the potential role of genetic variation in host-pathogen interactions of baculoviruses will be discussed.
HTML
-
Most of the work with respect to genetic variation of NPV has been done on those species infecting lepidopteran larval populations, ie. Alphabaculoviruses. For example, several studies of geographically dispersed populations of forest tent caterpillar (FTC), Malacosoma disstria, a lepidopteran pest of mixed forest systems in North America, have identified NPV isolates with distinct REN profiles suggesting that mulitple strains of this NPV occur (14, 16, 29). Figure 1 shows a REN profile of genomic DNA from a Malacosoma disstria NPV (MadiNPV) isolate, MadiNPV-A94, derived from a pooled larval sample from a single FTC population. The MadiNPV-A94 DNA profile shows a number of submolar bands indicative of a mixed population of genotypes. Seven distinct genotypes were evident amongst the first 20 clones derived by plaque assays of MadiNPV-A94 in the M. disstria cell line Md203 (Fig. 1) (16). These pick plaque strains showed a range of infectivity and virus replication kinetics in three different M. disstria cell lines. Ebling and Kaupp (14) also characterized six geographic isolates of MadiNPV from different geographic populations of FTC. Although all had similar REN patterns, they were distinct and three contained multiple genotypes based on the presence of submolar REN bands. One of these isolates was significantly more infectious for 3rd instar FTC larvae than the remainder. Numerous other studies of NPV associated with lepidopteran pests of forest systems have shown similar trends with multiple strains of NPV readily isolated from different geographic populations. Several studies used single larval collection strategies to isolate genotypic NPV variants; for example, Malacosoma californicum pluviale NPV isolates were derived from single larva collections of isolated geographic western tent caterpillar populations and 14 distinct genetic variants based on REN profiles were detected among 48 isolates (6). A “single larva” collection strategy for surveying NPV genetic variability probably increases the chances of quantifying the genetic diversity present in a virus population as compared to analyses of isolates composed of pooled groups of larvae (18). For example, Graham et al. (20) surveyed winter moth, Operophtera brumata, populations in the Orkney islands and detected Operophtera brumata NPV (OpbuNPV) in 11 of 13 populations examined. They characterized NPV from 200 individual cadavers and identified 26 genotypic variants and in only one larval isolate were submolar REN fragments detected in analysis of the NPV DNA. This indicates that the individual larval sample unit increases the changes of detecting variants but it is still possible to detect individuals infected with multiple NPV strains in wild populations.
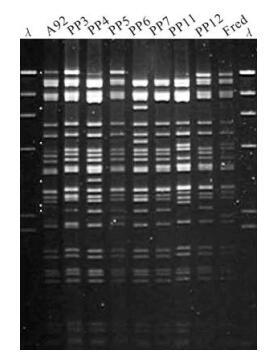
Figure 1. Restriction endonuclease (EcoRI) fragment profiles of Malacosoma disstria nucleopolyhedrovirus (MadiNPV) plaque isolates derived from MadiNPV-A92 including geographic isolates MadiNPV-A92 (Alberta, Canada) and MadiNPV-Fred (Fredericton, New Brunswick, Canada). Lambda phage DNA digested with Hind Ⅲ (λ) was used as size marker. Restriction endonuclease fragments marked with asterisk indicate those present in submolar ratios. (Adapted from references 16).
Similar survey strategies have identified genotypic variants and strains of alphabaculoviruses from major agricultural lepidopteran pests including: Mamestra brassicae NPV (MbMNPV) (3, 54); MacoNPV-A (15, 34); Spodoptera frugiperda NPV (SfMNPV) (46); Spodoptera exigua NPV (SeMNPV) (41); Autographa californica NPV (AcMNPV) (32, 57) and Helicoverpa armigera NPV (HearNPV) (27, 44). Numerous other examples could be cited but these serve as examples of the range of genotypic diversity found in field populations of baculoviruses.
-
There is less data available on the genetic diversity in betabaculovirus species, but where multiple isolates have been characterized there is also evidence for genetically distinct strains among geographic or temporal GV isolates. Crook et al. (11) examined seven geographic isolates of Cydia pomonella GV (CpGV) from around the world and found very little variability based on REN profiles. Most CpGV isolates were the same as the Mexican isolate (CpGV-M) and these differed only minimally from an isolate from Russia (CpGV-R) and an in vivo cloned strain from England (CpGV-E2). Recently, Eberle et al. (13) used RFLP analysis and partial genome amplification and sequencing to characterize eight new field isolates of CpGV. The most abundant genotypes in these isolates were identical to the previously describe CpGV-M, -R and -E2 variants; however a fourth genotype was identified. These authors conclude that these four major genotypes describe the genetic diversity of CpGV and that some geographic isolates appear to contain different mixtures of these genotypes. In contrast to CpGV, Crook (10) identified at least 11 genotypic variants based on REN profiles from among 15 geographic isolates of Artogeia rapae GC (ArGV) and these fell into three subtypes based on the continent of origin. In only two ArGV isolates was there evidence of submolar REN bands suggestive of multiple genotypic variants within a single geographic isolate. Similarly, Vickers et al. (55) were able to define three genotypic variants among eight geographic isolates of Phthorimaea operculella GV (PhopGV) that infects the potato tuber moth. Although GV are often considered to have a very narrow host ranges, Goto et al. (19) showed that GV isolated from six noctuid species were variants of Xestia c-nigrum GV (XecnGV) and the XecnGV isolates from each host species represent unique geneotypic variants with minor changes in REN profiles due in part to small insertion/deletions and single nucleotide changes within REN recognition sites. Each of the XecnGV variants was infectious for X. c-nigrum larvae.
Nucleopolyhedrovirus
Granulovirus
-
Many of the isolates described above are uncloned field isolates of baculoviruses. Despite being uncloned many of the distinct isolates appeared to have limited genetic heterogeneity at least as detected by RFLP analysis (ie. no submolar populations of REN fragments were detected). This is the case particularly for those isolates which were derived as single-infected-larva isolates. However, there are a few notable exceptions; Graham et al. (20) found one OpbuNPV isolate with submolar REN bands out of 200 single-infected-larva isolates analyzed, and an apparently single-infected-larva isolate of PaflNPV contained 24 genotypic variants (9). Even among those baculovirus isolates that appear to consist of a single dominant genotype cloning efforts have demonstrated otherwise. Many baculovirus isolates; for example, AcMNPV (32, 57); HearNPV (18, 44); LdMNPV (37); HycuNPV (28); and SfMNPV (30, 47) have been examined using in vitro plaque purification in susceptible insect cell culture lines whereby the BV recovered from single plaques are the progeny of single infecting BV or genotype. However, it has been argued that the in vitro cloning method which selects only from BV infections in cell culture may not appropriately reflect genotypic variation in original wild type isolates (9). This view is supported by the high frequency of deletion viruses selected within in vitro cloning studies with AcMNPV (45), SeMNPV (40) and SfMNPV (47, 48). A number of plaque purified virus strains from these studies contained large deletions and were unable to infect insect hosts per os as a result of the deletion of one or more of the baculovirus pif genes which are essential for oral infectivity. Thus, Cory et al. (9) suggested that the in vivo cloning strategy of Smith and Crook (49) is a more appropriate approach for determining the spectrum of genotypic variation in natural populations of baculoviruses. However, even in this case where only baculovirus strains that are infectious per os are selected, the results need to be interpreted with some caution particularly for multiple nucleopolyhedrovirus (MNPV). It has been estimated that MNPV ODV may contain between 1 and 29 genomes and that multiple genotypes can be co-occluded within a single ODV and OB (1, 4). Thus it is possible that minor genomic variants, even those that are defective with respect to per os infectivity, can be maintained within a heterogeneous baculovirus population presumably by the process of co-occlusion (48).
-
The genotypic variation in baculovirus populations noted above has been associated with differences in phenotypic characters such as infectivity (LD50 and LC50 estimates), pathogenicity (speed of kill-ST50 & LT50 estimates), virus productivity and host range. The variation in infectivity of different field strains of a baculovirus typically ranges up to 10 fold. For examples, the LD50 for HearNPV isolate G4 is approximately 3X higher than that of the C1 isolate when tested in dose response bioassays with 3rd instar H. armigera larvae (58). Similarly, MacoNPV-A isolate 90/2 is approximately 10X more infectious than geographic isolate 90/4 in 1st instar M. configurata larvae (34). However, much more significant differences in infectivity have been noted among plaque purified strains of African isolates of HearNPV; clonal strain HearNPV-NNgi had an LD50 of only 10 OB/3rd instar H. armigera larva compared to 3115 OB for the better characterized G4 isolate. Speed of kill estimates need to be interpreted carefully as it is essential that biologically comparable doses be used for the isolates being compared. However, where careful bioassays have been conducted LT50 have been shown to differ significantly. For example, Cory et al. (9) found that PaflNPV isolate Pf16 took 6 days longer to kill larvae than did the fastest isolate Pf12. In many cases in vitro cloned baculovirus strains have been shown to kill host larvae in half the time required for the comparable field isolate from which they were derived. For example, Harrison et al. (21) found the LT50 for 4 of 6 plaque isolates of SfMNPV ranged from 37.4-49.7 hours versus 80.5 to 90.5 hours post-infection for the field isolates from which they were derived. The other two SfMNPV plaque isolates were not infectious per os. Interestingly, these SfMNPV plaque isolates had genomic deletions of variable lengths but all had the egt gene deleted thus explaining the reduction in time to kill. There is also evidence for differential host range or at least differential efficacy among baculovirus isolates when their infectivity and pathogenicity is examined in alternate insect hosts. For example, Kolodny-Hirch and van Beek (31) showed that when a wild-type AcMNPV isolate was subjected to 20 passages through diamondback moth, Plutella xylostella, larvae its infectivity for P. xylostella increased significantly with the LC50 value of AcMNPV-Px20 being 15 times lower than for the wt-AcMNPV. These experiments were undertaken with uncloned virus isolates and REN analysis indicated there was an increase in prevalence of specific submolar REN fragments suggesting that there was a shift in the frequencies of particular genotypes in the AcMNPV-Px20 isolate. Similarly, Hitchman et al. (22) demonstrated that the proportion of various genotypes within PaflNPV differed upon passage through different alternative hosts. These authors suggest that the selection of different genotypes in different host species may play a role in maintaining genetic diversity in baculovirus populations. Finally, Hodgson et al. (23) provided evidence that specific PaflNPV isolates may have higher infectivity than others depending on the species of food plant the host insect consumes.
-
There are currently several baculovirus species for which the complete genome sequence is available for more than one strain or isolate. Although these complete genome sequences by no means reflect the total diversity of genomic difference within baculovirus species they do reveal some interesting trends (Table 1).The MacoNPV-A isolates 90/2 and 90/4 were originally derived from M. configurata populations in close geographic proximity and during the same temporal pest outbreak. The complete sequence of these isolates revealed that the MacoNPV-A 90/4 strain (AF539999) had 99.5% identity to strain 90/2 (NC_003529) but also had 521 single nucleotide substitutions and numerous small insertion/deletions relative to 90/2 (33, 34). The major region of divergence between the two genomes occurs between hr1 and orf31, a region that in 90/2 contains three baculovirus repeat ORF (bro) genes and one of these, bro-a, is absent in strain 90/4. This region which accounts for only 7.7% of the genome contains 50% of the total nucleotide changes in 90/4 relative to 90/2. This along with evidence from the genome sequence of the closely related MacoNPV-B species indicates that this region and particularly the bro-a, -b and -c genes are hot spots for sequence variation and gene rearrangements in the NPVs from M. configurata (33). Despite the high level of DNA sequence identity there are 49 ORFs that have 1-12% amino acid variation between the two strains and in addition there numerous differences in promoter motifs for many genes and substantial differences in hr elements except for hr2. While genome sequence analysis has not as yet provided definitive answers as to why 90/4 is less infectious than 90/2 a number of avenues for further investigation have been identified (34). Although the MacoNPV-A 90/4 strain was plaque-purified, the genome sequence assembled from shotgun clones clearly showed 214 polymorphisms of which the majority occurred within ORFs and there are 47 amino acid polymorphisms spread through 26 genes. It is possible that such a pool of sequence polymerphisms within in a population of a virus strain is an efficient mechanism for maintaining adaptability to changing environments and therefore an evolutionary advantage.

Table 1. Comparison of Complete Genome Sequence of Variant of NPV Species
The field isolate from which 90/4 was plaque purified was a heterogeneous population as determined by RFLP analysis which showed substantial populations of submolar fragments (34). In many previous genome sequencing studies no attempts were made to determine whether a plaque purified strain was representative of the population from which it was derived. In this study, numerous clones of selected REN fragments derived from DNA preparations from the original MacoNPV field isolate were sequenced and several genotypes including that of the 90/4 strain were identified. The MacoNPV-90/4 genotype represented approximately 25% of the mixed genotype population from which it was derived. The 90/2 genotype also appeared to be present in this mixed genotypic population of MacoNPV-A.
The genomes of two HearNPV strains, C1 (NC_ 003094) and G4 (NC_002654), have also been sequenced and compared in detail (5, 58). These strains were both isolated from H. armigera populations in Hubei province, China but as stated above the isolates have different virulence traits. HearNPV-C1 is 644 bp smaller than strain G4 and has 555 bp substitutions relative to G4 with the vast majority of these occurring in coding regions and 183 amino acid variations occur in 51 ORFs. This level of variability is similar to that observed between the two MacoNPV-A strains. The major differences between HearNPV C1 and G4 were in the bro-b gene and hr1, hr4 and hr5 not unlike the situation in MacoNPV-A. Zhang et al. (58) suggest that because the hr regions have been implicated in baculovirus regulatory processes as transcriptional enhancers and potential origins of DNA replication the observed alterations and recombination in these regions may explain the differences in temporal pattern of replication of G4 versus C1 virus that has been observed in cell culture lines.
The genome sequences of two isolates of AnpeNPV which infect the Chinese oak silkworm, Antheraea pernyi, have been completed (17, 43). A field isolate, AnpeNPV-Liaoning Z strain (NC_008035), and a pick-plaque purified strain AnpeNPV-L2 derived from the Lianoning isolate (EF207986) have 99.9% DNA sequence identity. However, the AnpeNPV-LZ genome is 384 bp longer due largely to insertions in intergenic regions and higher numbers of repeats in hr regions. Some of the most significant differences between the two strains include a truncation of egt in the LZ strain (79 aa vs 132 aa for the L2 strain) and 25 bp sub-stitutions in the putative desmoplakin and DNA polymerase genes that create frame shifts and slight truncations of these proteins (17). Interestingly, a sliding window analysis of nucleotide divergences between the two genomes indicated the differences were not random (17) but the regions of highest divergence did not include the hr regions or bro genes as was the case for MacoNPV and HearNPV isolate comparisons. Unfortunately, not much is known about the biological differences between the AnpeNPV strains so the relevance of the genetic diversity is not clear (17).
Two SfMNPV strains have recently been sequenced: SfMNPV-19 (EU258200) a virulent geographic isolate from Brazil (56) and SfMNPV-3AP2 (NC_009011) a plaque-purified strain derived from a field isolate from Missouri, USA that has a fast killing phenotype (21). Analysis of SfMNPV-19 DNA sequence that was derived from two or more REN cloned fragments suggest the number of polymorphisms is very low (0.12%) in this isolate (56). The latter investigators undertook a Clustal W alignment of the genomes of the two SfMNPV strains and the results confirmed that these are variants of the same virus species. The SfMNPV-3AP2 genome is 1235 bp smaller than SfMNPV-19 largely due to a 1427 nt deletion in 3AP2 which results in the loss of a portion of the 3' end of egt and the 5' of SfMNPV-19 ORF26. In addition, the putative dUTPase gene in SfMNPV-19 is truncated compared to the homologue in 3AP2 (56). The SfMNPV-3AP2 clone was one of a series of plaque isolates derived from six individual-cadaver-isolates from which the SfMNPV DNA was purified and used to transfect Sf21 cells. One plaque was selected from each of the single-cadaver-isolates and insect bioassays indicated that all but two were significantly more virulent than the parental field isolates (21), and the remaining two strains were not infectious per os. Harrison et al. (21) sequenced the egt region of each of the plaque purified strains and all had significant deletions ranging from the 1427 nt deletion in 3AP2 to an almost 12kbp deletion in SfMNPV-4AP2 that starts in the 3' end of lef-7 and stops near the 3' end of pif 2. The deletion of a functional pif 2 gene product in two of the plaque isolates probably explains their lack of oral infectivity. Although Harrison et al. (21) indicate there is no evidence that egt deletion mutants have a selective advantage in cell culture infections, it is remarkable that all the plaque isolates analyzed have the egt deletions. The genomic alteration in these SfMNPV plaque isolates are very similar to those described earlier for nine plaque-purified strains from a mixed genotype Nicaraguan isolate of SfMNPV (47, 48). The REN profiles of all the SfMNPV-NIC strains were analyzed and eight were shown to have deletions and sequence analysis of cloned REN fragments confirmed that all lacked a functional egt and several had large deletions encompassing a region that includes pif-2 and pif-1 in the parental genome. These authors demonstrated that mixtures of SfMNPV-NIC genotype B, a plaque strain representing the entire genome, plus specific ratios of certain deletion genotypes were more virulent than genotype B strain alone (48). These results suggest that retaining the mixed genotype populations that occur in natural field isolates may have an advantage for developing bioinsecticides based on baculoviruses (48).
In contrast to the SfMNPV in vitro selected plaque isolates cloned from wild-type SfMNPV field isolates, SeMNPV variants have been cloned from the wild-type isolates SeMNPV-US (Florida, USA) and SeMNPV-SP2 (Spain) using an in vivo cloning approach (41). In this case numerous rounds of in vivo cloning were required to produce pure and stable genotypic populations. The predominant region of variation in these genotypes was mapped to the hr1 region and there were differences in the number of repeats within hr1. In contrast to the increased virulence associated with some of the deletion mutants and mixed genotype populations of SfMNPV variants, the SeMNPV variants acted as parasitic genotypes that persisted and reduced the virulence of SeMNPV isolates (42).
-
Several of the mechanisms alluded to above have been suggested for the generation and maintenance of genetic diversity in baculovirus populations (53). Several of these mechanisms have particular relevance for genetic diversity within baculovirus species. Firstly, the complete genome analysis of isolates or strains within a number of NPV species confirm earlier data that suggested that hr and bro gene regions are hot spots for intragenomic recombination and variation. For example, complete genome data from MacoNPV-A and HearNPV strains support the suggestion that regions containing bro genes and hrs are hot spots for intragenomic recombination and variation. This has similarly been suggested for BmNPV strains (36).
In a few cases variation in hr and putative non-hr origins of replication, particularly with respect to the length and number of repeats, has been postulated to be related to the mechanism of baculovirus DNA replication, i.e., rolling circle mechanism (53). It has been observed during baculovirus replication in cell culture that hr and non-hr origins of replication are often duplicated and baculovirus genomes with multiple copies of these sequences may have a selective advantage over the wild-type genotype during replication in cell culture. This mechanism may also be involved in the rapid generation of deletion mutants that are selected for upon repeated passage in cell culture lines as for example in AcMPNV (45) and SfMNPV (47). The potential significance of this postulated mechanism in the evolution of baculovirus genomes among field isolates replicating in insects is not yet clear.
There is also ample experimental evidence that genetic elements can be interchanged between baculovirus genomes by homologous recombination when variants of baculovirus species or two species of closely related baculoviruses co-infect a common host in vitro (38) or in vivo (25). There is also evidence from genome analysis of MacoNPV-B that suggests a recombination event between a MacoNPV virus and Xec-nGV virus possibly involving homologous recombination between bro gene homologues in the two viruses (33).
Finally there are many examples suggesting transposable elements play a role in recombinant between baculoviruses and host insect genomes (2). The majority of transposable elements inserted into baculovirus genomes do not contain open reading frames per se but some have been associated with few polyhedra phenotype as result of their insertion into the fp25K gene disrupting the normal open reading frame.

 DownLoad:
DownLoad: