











-
Canine parvovirus type 2 (CPV-2) is a highly contagious infectious agent that causes gastroenteritis in dogs. CPV-2 emerged in 1978 as a new dog disease agent and rapidly spread around the world in domestic and wild dogs causing high morbidity (100%) and frequent mortality (up to 10%) [1]. In the 1980s, two antigenic variants of CPV-2, distinguishable using monoclonal antibodies (MAbs), emerged almost simultaneously and were termed as CPV-2a and CPV-2b respectively [21]. Currently, CPV-2a is the prevalent field strain in Italy and Germany, while CPV-2b is common in USA, Taiwan and Japan [2, 14]. CPV-2c having a change of D426E occurring in a strategic residue responsible for the antigenicity of CPV-2b has been detected in Vietnam, Italy, Spain, Germany, United Kingdom and South America [7, 17]. CPV-2 was isolated for the first time in India by Ramadass and Khadher in 1982 [24] and after that several incidences of the disease have been reported from different parts of the country. The prevalence of CPV-2a has been documented in 2001 in India by Narayanan et al [20]. It was also reported that CPV-2b is more common when compared to CPV-2a in Northern India [19]. The prevalence of CPV-2c has also been reported in India by Nandi et al [19].
The laboratory diagnosis of CPV-2 infection mostly relies on detection of virus in faecal samples or antibody in serum samples of suspected animals. The antibody in the sera can be detected preferably by competition ELISA (c-ELISA) and sometimes by agarose gel immunodiffusion (AGID) but unable to differentiate vaccinated from infected animals. Considering, the extreme severity of the disease, the prompt, sensitive and accurate diagnosis of the canine parvoviral disease on the basis of antigen detection is of paramount importance. It can be done by demon-stration of viral antigens in the faecal samples by AGID, virus isolation in cell culture, CIE (counter immunoelectrophoresis) test [24], virus neutralization test, haemagglutination (HA) test, immunoelectron microcopy, immunofluorescent test, antigen-capture (sandwich) ELISA or genomic DNA detection by polymerase chain reaction (PCR) [15, 30]. Recently, the PCR technique has been increasingly used as a tool for the diagnosis of several viral infections. Further, real-time PCR [6], nucleic acid hybridization or dot blot, in situ hybridization [5], and loop-mediated isothermal amplification (LAMP) [10] etc with varying degree of sensitivity and specificity have been employed for diagnosis of CPV-2.
In India, the disease is widely prevalent and there exist about 25 million dogs with a high proportion of stray dogs capable of harbouring the CPV-2 without showing any symptoms. Since copious amounts of CPV-2 are shed in the faeces of infected dogs and the infection is easily spread among susceptible animals through faecal-oral route, rapid and early diagnosis of CPV-2 lead to proper treatment with high rate of recovery. The AC-ELISA for detection of viral antigens is an ideal choice as it is less time-consuming, easy to perform, having good sensitivity and specificity and well suited for screening of large number of samples. There is no indigenous ELISA kit available in India for CPV diagnosis and expensive ELISA kits are available only in few countries. So, the present study was undertaken to develop a polyclonal antibody (PAb) based AC-ELISA for detection of CPV antigens from faecal samples of infected dog and infected cell culture supernatant [4, 8, 12, 16, 18, 23].
HTML
-
A total of 129 faecal samples were collected or received at Virus Laboratory, CADRAD, for diagnosis from dogs suspected of CPV-2 infection. The rectal swabs were taken from individual dogs suspected of CPV-2 infection and suspended (in the ratio 1:9) in Hank's balanced salt solution (HBSS) containing streptomycin (100 mg/L) and penicillin (100 000 IU/L). It was filtered through a disposable syringe filter (0.45 μm) (Millex, Milipore, USA) and then centrifuged at 10 000 r/min at 4℃ for 5 min in a refrigerated centrifuge. The supernatant was carefully pipetted out and stored at -20℃ till further use [18].
-
MDCK cell line was obtained from National Centre for Cell Science, Pune. It was maintained at the Virus Laboratory, CADRAD by using Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) with 10% fetal calf serum (FCS) as growth medium. Gentamicin was added in the medium at the rate of 50 mg/L (Life Technologies). The MDCK cell line was sub-cultured and grown to 70% of cell culture flask, 0.5 mL of processed faecal samples were added and incubated for 1 h at 37℃. After incubation, the infected cell monolayer was washed three times with DMEM and 5 mL of DMEM medium with 2% FCS was added. The infected cells were incubated at 37℃ for 3-5 days.
-
Purification of CPV was carried out according to the protocol described by Teramoto et al. with some modifications [30]. Infected cells were disrupted and then centrifuged at 3 000×g for 30 min at 4℃ to obtain cell pellet. The pellet was resuspended in PBS, sonicated at 30 μ amplitude for 1 min to disrupt the cells, centrifuged at 8 000×g for 10 min and supernatant was collected. The supernatant was treated with 8% PEG-6000 and kept on the magnetic stirrer overnight for precipitation of the virus. The PEG-treated supernatant was pelleted by centrifu-gation at 6 000×g for 30 min and the pellet was re-suspended in PBS for ultracentrifugation. Two mL of the resuspended virus was then carefully layered over discontinuous gradient of 60% and 40% sucrose in 12 mL centrifuge tube (4.5 mL 60% sucrose + 4.5 mL 40% sucrose) and centrifuged at 100 000 ×g for 2 h at 4℃ in an ultracentrifuge (Sorvall, Ultracentrifuge, swinging rotor AH629, USA). At the interface of sucrose gradients thick opaque band was observed. The band containing the virus was collected, diluted with equal amount of PBS and again centrifuged at 100 000×g for 2 h at 4℃ in an ultracentrifuge (Sorvall, Ultracentrifuge, USA) using fixed rotor.
-
Hyperimmune serum was raised in rabbits and guinea pigs by inoculating purified virus for three times over a period for 28 days. One hundred μg of purified virus particles in 0.5 mL of PBS along with 0.5 mL of Freund's complete adjuvant (FCA) was inoculated subcutaneously in rabbits. Similarly, 50 μg of purified virus particles in 0.5 mL of PBS along with 0.5 mL of FCA was inoculated subcutaneously in guinea pigs. Inoculation was repeated after 14 and 28 days with Freund's incomplete adjuvant (FIA) with same amount of virus particles. The test bleeding was done 7 days after the third immunization and the presence of antibody was detected by AGPT.
-
Positive control antigen was prepared from MDCK cells grown in 75-cm2 tissue culture flask to a near confluent monolayer and infected with CPV-2. At 96 hours post-infection (hpi), the virus was harvested and centrifuged at 3 000×g for 30 min. The pellet was resuspended in PBS, sonicated at 30 μ amplitude for 1 min and cell debris was removed by further centrifugation at 8 000×g for 10 min, the supernatant was taken and aliquoted for further use. Negative control antigen was prepared in the similar manner from uninfected MDCK cells and supernatant taken.
-
In the AC-ELISA, the rabbit hyperimmune serum and guinea pig hyperimmune serum were used as capture antibody and tracing antibody respectively. ELISA plates (Maxisorp Nunc, Denmark) were coated with the capture antibody in carbonate-bicarbonate buffer, pH 9.6 and kept at 37℃ with continuous shaking for 1 h. The unbound capture antibodies were removed by five washing with washing buffer (PBS containing 0.05% Tween-20) and free sites were blocked by adding blocking buffer (PBS with 1% BSA). After incubation and washing, the test antigen (diluted 1:2 in blocking buffer) was added, incubated at 37℃ for 1 h and washed five time followed by addition of tracing antibody to each well. After incubation at 37℃ for 1 h, the plates were washed five times with washing buffer followed by addition of 100 μL of 1:2 500 dilution goat anti-guinea pig horseradish peroxidase (HRPO) conjugate (Sigma, St. Louis, USA) to each well. The plates were incubated at 37℃ for 1 h, washed five times with washing buffer and freshly prepared substrate/chromogen mixture [4 mg Orthophenylene diamine (OPD) in 10 mL substrate solution containing 2.5 μL of 3% H2O2 solution] was added to all wells. The plates were kept for 15 min in the dark at room temperature and the color reaction was stopped by adding 50 μL of 1 mol/L H2SO4.The plates were read at 492 nm wavelength on an ELISA reader (BioRad 680 Microplate Reader). A value twice or more than the mean OD value of the negative antigen control was considered as the positive/negative cut-off value (i.e. P/N ratio≥2).
-
The primer set pCPV-2ab (F) 5'-GAAGAGTGGT TGTAAATAATT-3' (21-mer) and (R) 5'-CCTATA TAACCAAAGTTAGTAC-3' (22-mer) used to amplify the part of the VP2 gene of CPV from nt 3 025 to 3 706 was custom synthesized to yield a PCR product of 681 bp[22]. The PCR reaction mixture consisted of 5 μL of template DNA, 5 μL of 10×Taq buffer (containing 15 mmol/L MgCl2), 1 μL of each of forward primer and reverse primer (20 pmol), 1 μL of dNTPs mix (10 mmol/L each), 1 μL of Taq DNA polymerase (1U/μL) and nuclease-free water up to 50 μL. Amplification was performed in a thermocycler (Applied Biosystems) and the cyclic conditions consisted of initial denaturation at 95℃ for 5 min followed by 30 cycle of 95℃ for 30 sec, 55℃ for 1 min and 72℃ for 1 min and a final extension at 72℃ for 10 min [18]. At the end of PCR, the amplified products were analyzed on 1.0% agarose gel containing ethidium bromide to a final concentration of 0.5 μg/mL. 10 μL of amplified product was mixed with 2 μL of bromophenol dye (6×) and loaded into the well and run along with 100 bp DNA ladder in 1×TAE electrophoresis buffer at 5 V/cm2 and the progress of mobility was monitored by migration of dye.
-
A total of 152 samples including faecal samples (129) and CPV-infected cell culture supernatant of different passages (23) were tested by AC-ELISA and PCR. Results of AC-ELISA were compared with PCR results and relative sensitivity, relative specificity and accuracy of the AC-ELISA were calculated.
Processing of fecal samples
Virus isolation
Purification of CPV
Production of PAb
Positive and negative antigen preparation
Optimization of AC-ELISA
PCR Assay
Testing of samples and analysis of data
-
Three faecal samples found positive in PCR were subjected to blind passages in MDCK cells. In the first 3 passages, no cytopathic effects (CPE) were seen. However, from 4th passage onwards, MDCK cells exhibited CPE characterized by rounding of cells, granulation and aggregation of cells after 72 hpi which increased subsequently and widely distributed in whole monolayer (Fig. 1). The results are in accordance with the Joshi et al [11]. Then the virus was harvested and freeze-thawed thrice and stored at -20℃ until further use. The virus titre was determined by Reed and Muench method [25]. The titre of virus was calculated from three different readings and the mean was found to be 104.8 TCID50/mL. Using this as inoculums, 2 000 mL of CPV-2 infected cell culture fluid was produced for purification of virus.
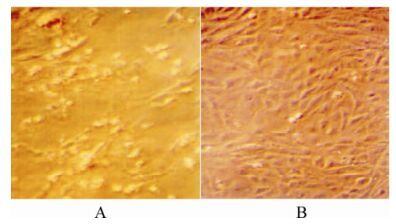
Figure 1. MDCK cells infected withCPV-2 showing CPE after 96 hrs pi (A) and Confluent monolayer of MDCK cells (B).
-
In the sucrose density gradient ultracentrifugation, the pellet obtained was resuspended in 2 mL of PBS. The protein content of the purified virus was estimated by U.V. method in nanodrops spectrophotometer (Thermo-scientific, USA) and found to be 350 μg/mL. After test bleeding, the presence of antibody was evaluated by AGPT and a distinct band was visualized between the raised hyperimmune serum in both species and purified virus (Fig. 2). Final bleeding was done after 10 days of the final immunization, serum collected, inactivated and kept at -20℃ for further use.
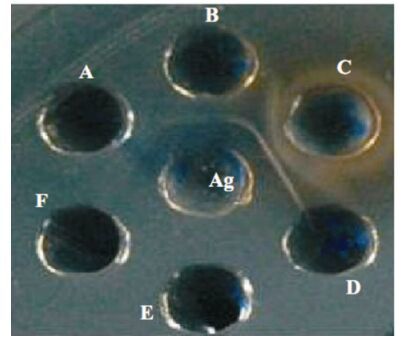
Figure 2. AGPT showing visible precipitin band with CPV antigen in central well. A, Negative anti serum B, Hyperimmune serum raised in guinea pig. C, Hyperimmune serum raised in rabbit.
-
The optimum dilutions of capture and tracing antibodies were selected by check-board titration against fixed dilution/concentration of CPV antigens with different dilution of rabbit and guinea pig hyperimmune serum and most satisfactory result was obtained at a 1:1 600 dilution for the capture antibody and a 1:400 dilution for the tracing antibody with 1:2 dilution of the positive antigen and the negative antigen. At this dilution the total protein content of the positive antigen control was about 1.0 μg per well and the P/N ratio was > 2.5. These dilutions of reagents were followed throughout the study and subsequently for testing different samples. No excessive variations ( < 10%) between plates were detected and found statistically significant and found to be corroborated with Maree and Paweska [13]. The AC-ELISA was able to detect different variants of CPV-2 such as CPV-2a, CPV-2b and CPV-2c. It detected various CPV-2 variants in the infected cell culture supernatant as positive and showed no reactivity with healthy MDCK cell culture derived negative control. Analytical specificity was confirmed for other canine enteric viruses such as canine coronavirus, canine adenovirus etc. and found to be nonreactive. The CPV-2 variants used in the study were typed previously on the basis of PCR and sequence analysis and maintained in the Virus Laboratory, CADRAD[19]. The analytical sensitivity of the AC-ELISA was determined using serial 10-fold dilutions of CPV-2 infected cell culture supernatant of known titer (104.8 TCID50/mL) and the detection limit of the AC-ELISA assay was up to 10-2dilution, equivalent to 102.8 TCID50/mL. The analytical sensitivity of the AC-ELISA was compared with the pCPV-2ab primer set based PCR [22] and detection limit of the PCR was up to 10-4dilution, equivalent to 100.8 TCID50/mL (Fig. 3). So, PCR was found to be about 100 times more sensitive than the AC-ELISA for detection of CPV in cell culture system (Table 1).
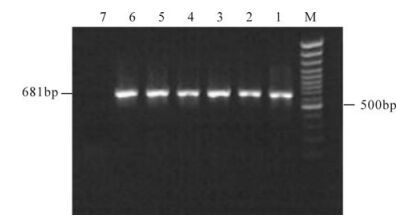
Figure 3. Agarose gel showing amplicon of 681 bp of CPV-2 using pCPV-2ab (F & R) primer. M, 100 bp DNA marker; Lane 1 to 5, 681 bp PCR products of samples 1 to 5; 6, 681 bp PCR product of positive control; 7, Faecal sample of healthy dog as negative control.

Table 1. Relative sensitivity of AC-ELISA compared to PCR for detection of CPV-2.
-
At the end of the electrophoresis, the gel was visualized under the UV transilluminator and an amplicon of 681 bp of VP2 genes was seen from positive faecal samples and positive control, but not in fecal sample from healthy dog (Fig. 4).
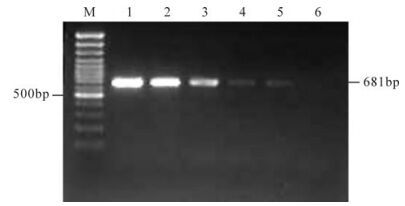
Figure 4. Sensitivity of PCR for detection of CPV-2. DNA was extracted from different dilutions (lane 2-6: 10-1 to 10-5, ) using primers pCPV-2ab DNA bands of expected size (681 bp) were visualized up to 10-4 dilution which is equivalent to a titer 100.8TCID50/mL. M, 100 bp DNA ladder.
-
In s-ELISA, 69 out of 152 samples were found positive, while in PCR 78 samples were found positive. Nine samples negative in AC-ELISA were found positive in PCR, 69 samples were positive in both AC-ELISA and PCR and 74 samples were found negative in both tests (Table 2). The relative sensitivity, specificity and accuracy of the AC-ELISA were found to be 88.4%, 100% and 91.4% respectively (Table 2).

Table 2. Relative performance of PCR and AC-ELISA for detection of CPV-2 in samples.
Virus isolation
Purification of CPV
Optimization of AC-ELISA
PCR Assay
Analysis of resultss
-
In the present study, an AC-ELISA for detection of CPV antigens was developed based on PAb raised in rabbit and guinea pig as capture and tracing antibody, respectively. The results of AC-ELISA were compared with PCR, a highly sensitive and specific method for diagnosing CPV. The relative sensitivity of AC-ELISA was found to be 88.4% indicating its high usefulness to identify PCR positive samples as positive. Good correlation between PCR and AC-ELISA results was found and corroborated with the findings of Bell et al[3]. However, the result of AC-ELISA depends on the conformational epitopes of the virus particles and antigen-antibody interactions whereas PCR can give positive results even in samples containing distorted virus particles. The relative specificity of AC-ELISA was 100% signifying the ability of the AC-ELISA to detect all the PCR negative samples as negative. Most of the ELISA developed for CPV-2 antigen detection was based on MAbs. Teramoto et al developed an ELISA test based on MAb for detection of CPV-2 in faecal samples and compared with HA assay and found a good correlation between the tests[30]. Rimmelzwaan et al developed double antibody sandwich ELISA based on MAbs for detection and quantification of CPV-2 antigen [26]. In India, Mohan et al used an imported ELISA kit based on MAbs for the detection of CPV-2 antigen in faeces and found more sensitive compared to classical HA test [16]. The sandwich ELISA has been found to be more sensitive compared to AGID and CIE [28].
The use of PAb as capture antibody has been shown to increase the sensitivity of s-ELISA compared to MAbs as capture antibody for diagnosis of bluetongue virus[9]. However, most of the ELISA developed for the diagnosis of CPV-2 was meant for detection of antibody [27, 29, 31, 32]. The advantages of AC-ELISA are easy to perform, less time-consuming and requirement of little amount of antigen and antiserum. The PCR-based diagnosis is beyond the feasibility of small diagnostic laboratories owing to its requirement of skilled manpower and costly equipments. Further, bile and other inhibitory substances present in the faecal samples of dogs make it necessary to extract DNA to carry out PCR and become a time-consuming process in case of large number of samples. However, the AC-ELISA can be very useful for diagnosis of CPV-2 in large number of faecal samples.
In conclusion, this study described a sensitive and specific antigen detection test for CPV-2, employing the PAb raised in different species using ultra-purified CPV-2. The AC-ELISA can be easily performed in the small laboratories and even at clinic level to obtain prompt, accurate and specific diagnosis of CPV-2, which can help in the effective and efficient management of the disease. Further, the same principle can be utilized to develop the dot-ELISA for the detection of CPV in the faecal samples of canines.

 DownLoad:
DownLoad: