











-
Ion channels control the flux of ions across the lipid membrane in an extremely sensitive and specific manner[102]. In 1964, Hoffmann and colleagues discovered the first anti influenza agent, amantadine, that target at the M2 proton channel [13], which led to an explosion in the number of identified virus ion channels and subsequent antivirus drug research.
Viral genomes encode a number of short auxiliary transmembrane subunits, which assemble into viral ion channels. Most of subunits are small peptides of about 100 amino acids which are just long enough to span the lipid bilayer once, for example, the M2 channel has 97 amino acids for each subunit [22, 23, 25]. These subunits assemble into homo or hetero oligomers, however most viarl ion channels are homo-oligomers, which form an internal water-filled pores. These kinds of channels or pores allow small molecules as well as ions such as protons and K+atoms to flux across the membrane, which may modulate both viral and host cellular activity [21].
Since ion channel has important functions in the virus life cycle, viral ion channels have become potential and attractive targets for antiviral therapy. This viral transmembrane protein has several unique properties that also make it ideal for the development of antivirals. In fact, scientists have proved that inhibitors of viral ion channels are efficacious in human infection, for example, antiviral drugs such as amantadine and rimantadine can inhibit the M2 channel of influenza A [6, 9, 41, 42, 44, 47]. However, one of the hurdles in the rational design of viral ion channel inhibitors is the lack of details about the ion channel structure. For example, how do these subunits assemble into homo or hetero oligomers and create the water filled pores? What are their structures? How do the ion channels gate and selectively function? And how can we use channel-forming proteins for antiviral therapy?
This review provides an overview of the current research on viral ion channels that is guided to some degree by the above questions. We mainly focus on the ion channels of viruses which can lead to serious infection in human beings and consider seven virus ion channels: M2 of influenza A, NB and BM2 of influenza B, CM2 of influenza C, Vpu of HIV-1, p7 of HCV and 2B of picornaviruses. These ion channels, a group of proteins belonging to the viroproins family, participate in viral functions including the promotion of release of viral particles from cells [26]. All these proteins can enhance the passage of ions and small molecules through membranes by variation of their concentration gradient [26].
HTML
-
The M2 channel is an essential component of the influenza A virus infectivity cycle, which is a homotetramer in its native state. M2 is encoded by a spliced mRNA derived from segment 7, which has two homodimers, each of the subunits containing 97 amino acids, that have their amino and carboxy termini directed towards the outside and inside of the virion respectively [38, 86, 88]. There are 19 residues that cross the membrane region which located between 24 amino acid, an N-terminal extracellular domain and 54 amino acid, C-terminal intracellular domain [50]. This homotetramer is composed of a pair of disulfide-linked dimers or a disulfide-linked tetramer [38, 60, 86]. The four TM domains of M2 are parallel to each other and form a left hand supercoil [72]. The M2 protein is posttranslationally modified by the formation of a intermolecular disulfide bond at Cys-17 and Cys-19 and by palmitorylation through a thioether linkage [38]. Pakmitylation works on Cys-50 to the cytoplasmic tail [39, 85, 96]. But a recent study discovered that palmitorylation of the influenza A M2 channel is not required for virus replication in vitro but partly contributes to the virus virulence in vivo [28]. The structure of the TM segment is an α-helix. A model for the raft has suggested that M2 has little associated with it. M2 can be raft anchored with its cytoplasmic domain because of palmitorylation site Cys-50. There may be other enveloped viruses that could encode such a protein with similar properties. Two possible gating mechanisms of the M2 channel have been proposed: (ⅰ) Gating. Electrostatic potential is involved in the mechanism. In high pH gradient, the side chain of residues His-37 on the four helices is in a fully deprotonated state which is oriented towards the lumen of the pore. However, in a low pH gradient, the side chain of four residues His-37 in a fully protonated state adopting interfacial position that does not exhibit occlusion of the pore [72]. (ⅱ) Flipping. The nitrogens in the ring are protonated by low pH. Because of the energetic state of the two nitrogens, the side chain flips around its Cα-Cβ bond [21, 45, 67]. In the the TM region, His-37 has been found to be involved in pH induced channel opening [97]. High proton selectivity is blunted by replacement of TM His-37 with Ala, Gly, or Glu [98]. Also, when water was replaced by deuterium oxide, a kinetic isotope effect was measured. Thus it possible that protons either bind to the histidine and are released or form short-lived proton "wires" across the His-37 barrier (Table 1) [56, 78].

Table 1. Properties of the putative ion channels of virus
Similarly, NB, BM2 is encoded by influenza B and and CM2 is encoded by influenza C. These proteins are type Ⅲ integral small membrane proteins and their channel protein subunits are all within a single transmembrane helix that, as a tetramer, forms a proton channel. They all have N-terminal extracellular domain and C-terminal intracellular domain motifs. The influenza B virus, like the influenza A virus, is endocytosed and uncoating occurs after fusion of the virion with the endosomal membrane [49]. Uncoating of the virus requires a low pH step. BM2 has little identity sequence compared with AM2, except for the HxxxW signature sequence for a proton channel [104]. Although they have low homology, the conductance is similar. The only palmitorylation site, Cys-50, is not present in BM2 [3]. The BM2 has a unique helical bundle which is purported to be a coiled coil, which is requires the SxxSxxxS motif (Ser-19, Ser-12 and Ser-16) [12, 104]. His-19 is supposed to serve as a selectivity filter. It is possible that the filter mechanism is just like M2 channel of influenza A virus. Protons that flow from the acidic medium are binding to the filter and finally released to the opposite side of histidine residue and then into the virion interior. A TM tryptophan residue is assumed to serve as an activation gate that is closed while the medium bathing the ectodomain is neutral or alkaline. The recent findings with the BM2 channel of influenza is assume that it is unlikely that NB channel is an ion channel of influenza B virus that cause acidification during uncoating. The NB protein is an integral membrane protein of influenza B virus, that contains 100 amino residues[19, 87]. This protein once was thought to be an indispensable ion channel of the virus, and was reconstituted and studied in bilayers. Chizhmaknov and Lamb, and Pinto both proved that the NB channel can exhibit selectivity for cation ions such as Na+ or H+ [19, 87] but this protein does not provide for acidification of oocytes.
The CM2 protein has many similar biochemical properties compared to the influenza A virus M2 and influenza B virus BM2 proteins, such as the NoutCin membrane orientation, the size of the ectodomain, the cytoplasmic tail and the formation of disulfide-liked oligomers [66]. It is also characterized as a tetrameric integral membrane glycoprotein, and the helices are tilted from the membrane by about β= (14.6 ± 3.0)°. Residue Met-65 is supposed to occlude the putative transmembrane pore [48, 87]. Together, these channel proteins from the three type of influenza constitute the first-known members of the single-pass proton channel family.
Virus protein "u" (Vpu) was discovered in the mid-1980s by Cohen and Strebel, respectively, and uniquely contributes to the virulence of HIV-1 infection of humans by enhancing the production and release of progeny virus particles [11, 83, 89]. The Vpu protein, which contains 80 to 82 amino acids (depending on the HIV-1 isolate) [18, 52], is of amphipathic nature and consists of a hydrophobic N-terminal membrane anchor proximal to a polar C-terminal cytoplasmic domain, and it requires the presence of lipids and water to adopt its native functional structure[18]. Two distinct domains of the Vpu allow it to perform multiple biological functions such that it can act as a channel and also interact with other proteins. Oligomerization of the Vpu TM domain results in the formation of sequence specific cation-selective channels [2, 61]. Although the secondary structure and tertiary fold of the cytoplasmic domain of Vpu have been determined by a combination of NMR, CD spectroscopy, and molecular dynamics calculations [51, 99, 100], the ion channel formation and gating mechanism is still unclear.
The hepatitis C virus (HCV), a major human pathogen associated with severe liver disease, and encodes a small membrane protein designated p7. The p7 polypeptide is composed of two long hydrophobic transmembrane domain connected by a conserved basic cytosolic loop [8]. Two topologies for monomeric p7 have been proposed: a double membrane-spanning hairpin topology in the endoplasmic reticulum with Its N and C-termini ends both facing the interior of the endoplasmic reticulum lumen [8, 63] and an L-shaped form with the C-terminus facing the cytoplasm[29, 43]. Because of the secondary structure prediction, it is proposed that the transmembrane domain is an α-helix[8]. Patargias et al. and Clarke et al. proposed that in the heptameric model of p7, the N-terminal helix lines the pore [10, 62]. Mutations in the cytosolic loop domain make the channel deactivate [32]. Modeling analysis indicates that a His-17 residue would be a pore-facing residue, which suggests that p7 may be sensitive to pH with respect to its function [62]; it is a cation-selective channel at normal pH with a measured conductance between ~86 and ~100pS [64]. In the absence of high resolution structure data, the channel formation and gating mechanism of p7 is not clear but it is necessary to establish which residues may line the pore to help interpret low resolution electron microscopy data.
Picornavirus proteins arise from a large polyprotein precursor that is cleaved by viral proteases. The 2B protein is one of the nonstructural proteins encoded by enteroviruses such as poliovirus and coxsackie virus [14, 59]. In the 9 distinct genera of Picornaviruses, only Coxsackievirus and Poliovirus 2B protein have been proved that they both belong to viroporin family[1, 14, 15, 62].2B is thought to consist of two hydrophobic transmembrane stretches which spans the membrane by means of an amphipathic helix [93, 94]. Highly conserved C-terminal TM domain reproduced the capacity of the full 2B protein to efficiently permeabilize bilayers made of anionic phospholipids. Insertion into lipid monolayers and circular dichroism determinations were consistent with penetration of the TM1 helix into anionic and zwitterionic membranes. One of the TM domains is rich in lysine, a short linker region followed by another TM domain [61, 94]. Mutations of the hydrophilic residues within the short linker region impair the multimerization and decrease membrane permeability, while mutating the Try residues toward the C-terminus of the second transmembrane domain also allows abrogation of membrane permeability without affecting mutimerization [1, 7, 59, 92, 94]. In the carpet-like mechanism, one of the TM domains lies on the surface of the membrane, and the other one forms the pore. This mechanism allows the permeation of molecules of a wide range of sizes. Using fluorescence resonance energy transfer microscopy and a two-hybrid system[53], it has been suggested that most of the 2B protein located in membrane regions oligomerize as dimers and tetramers [1]. The 2B protein forms a pore which is 6 in diameter [1] and allows the diffusion of molecules of MW under 1 000 kDa. Additionally, ions and small molecules can also pass through the pore. The ion channel formation and gating mechanism is still unclear.
-
The viral channels represent miniaturized channel systems, which play an important role in the viral life cycle.
The first line of evidence replicating the A/M2 protein in the acidification of the virion came from experiments with amantadine, which inhibits the "early" uncoating step in replication, this evidence has been reviewed [36]. In common with many other envelope viruses, after attaching to the host cell membrane, the influenza A viruses fuses with this section membrane to form a vesicle or endosome that only occurs in a narrow pH range from 5.0 to 5.5 (Fig. 1). When the His-37 senses the low pH in the endosome, the M2 protein conducts protons into the interior of the virion. Because of the lowering of the pH, the conformation of the M1 and HA proteins changes which eventually leads to the uncoating of the viral genome. During the late stage of the infectious cycle, the M2 protein is transcribed in the infected cell and maintains a near-neutral pH in the Golgi, which can make the HA protein adopted the high-pH conformation which results in the assembly of functional HA protein in the virion [84]. When ionophore monensine catalyzes the exchange of Na+ for H+ across membranes, the HA was inserted into the plasma membrane in its high pH form, suggests that the A/M2 protein and monensin both act to shunt the pH gradient across the Golgi membrane [27]. The BM2 ion channel from the influenza B virus and the CM2 ion channel from influenza C have a function similar to that of the M2 ion channel in so far as uncoating the virus at a low pH in order to allow dissociation of the RNPs from the matrix protein [49], but the pH modulating activity of the CM2 protein is substantially less than that of the M2 protein [4]. The CM2 protein forms a voltage-actived ion channel permeable to Cl-but not to cation [40]. Comparing the electrophysiological properties, the BM2 ion channel is similar to the M2 channel and the NB channel has some features in common with the CM2 channel [4, 57].
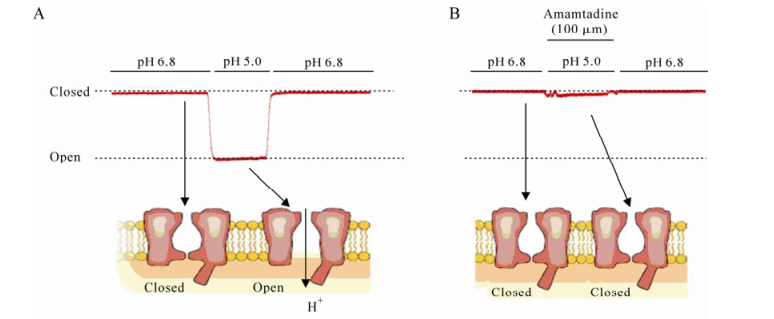
Figure 1. Whole-cell membrane currents of M2 ion channel. The pH off the bathing medium was lowered from pH 6.8 to pH 5.0. The membrane voltage was held at -60mV. A: M2 ion channels were activated by lower pH condition. B: M2 ion channel inhibited by 10 μm amantadine.
Unlike the ion channels from the influenza virus, as an accessory protein of HIV-1 Vpu presents two distinctive biological functions in HIV-1 life cycle [5]. Firstly, Vpu facilitates the budding of new virus particles from infected cells surface; without an active Vpu protein, newly formed virions accumulate in the cells increasing cytotoxicity as a result [46, 52, 55, 101]. This budding activity requires an intact TM α-helical domain [77]. The presence of a transmembrane helical domain from Vpu results in ion channel activity and may also affect the virion budding process. The cellular block to particle production in simian-human heterokaryons is relieved by the expression of Vpu [95]. These maybe means that a new host cell restriction of HIV replication is that is present in human cells and overcome by Vpu [95]. But the molecular mechanism by which Vpu facilitates virion release remains unclear [54]. Second, Vpu also enhances the degradation of CD4 in the endoplasmic reticulum that an activity associated with interactions between the cytoplasmic domains of Vpu and CD4 [82, 101]. Because of the absence of Vpu, processing of gp160 to form the gp41 and gp120 polypeptides is impaired since stable complexes with CD4 in the endoplasmic reticulum of infected cells [76]. Thus, Vpu indirectly regulated processing of gp160 in the trans-Golgi compartment into the gp120 and gp41 proteins needed for the assembly of infectious virus particles. To a lesser extent, enhancement of virion particle release is both dependent on the two highly conserved serine residues (Ser52 and Ser56) in the cytoplasmic domain which can be phosophorylated by caseine kinase-2 (Table 1) [37, 103]. The N-terminal transmembrane helix, which serves as a membrane anchor, is required to regulation virus secretion, most likely by formation of an ion channel[55]. These two functions of Vpu occur in separate subcellular location[77, 103].
Hepatitis C virus (HCV) infection is associated with chronic liver disease and currently affects about 3% of the world population [79]. Much research has been doing to elucidate the functions of HCV proteins and it has been determined that that the p7 protein forms a channel for conducting ions across the membrane [70, 79] and that it is essential for HCV infection but does not affect RNA replication [62].However, although in-vivo experiments show that the p7 protein is essential for infectivity in chimpanzee [69], By employing a novel HCV infection system and using a genetic approach to investigate the function of p7 in the viral replication cycle, it has been determined that p7 is essential for efficient assembly and release of infectious virions across divergent virus strains [35, 79]. It also has been demonstrated that the carboxy-terminal domain can function as a signal sequence that most likely promotes the translocation of NS2 into the ER lumen for appropriate cleavage by host signal peptidases [69]. Other observations indicate that p7 is primarily involved in the late phase of the HCV replication cycle and has a role in the secretary pathway [8] Thus, p7 is an important virulence factor that may modulate fitness [35, 79] and is also associated with virus persistence and pathogenesis. However, virus entry appears largely independent of p7 as the specific infectivity of released virions with a defect in p7 was not affected and in spite of the findings outlined above, the exact role of the HCV p7 protein in virus replication remains unclear
The 2B protein has been localized to the ER and Golgi complex, and to a lesser extent to the plasma membrane [14, 15, 72, 93], but is not transported outside the Golgi [14]. That the too large oligomers and lack of specific signals may be contribute the retention of 2B in the Golgi. Calcium is more permeable, thus it is postulated that the 2B protein may play an important role in infection, modulating apoptosis and lower molecular weight compounds by modulating calcium and proton or altering the solute balance across the plasma membrane[93, 94].
-
Inhibition of proteins which are essential in the virus life cycle is a basic approach in antiviral therapy. In ion channel target therapy, there are two general strategies (Table 1). Firstly, a small molecule or peptide drug inhibits the ion channel activity. Secondly, using anti-oligomerzation peptides prevent the formation of the pore [20]. The drug may either enter the pore [81]or diffuses into the hydrophilic/hydrophobic domain in the outer rim of the bundle[75]. Both small organic molecule and peptides can be used as a drug, but peptide drugs have two advantages over small molecule drugs. First, they are highly active and specific. Secondly, they accumulate in tissue to a lower extent and thus have reduced side effects and toxicity. But peptide drugs have very low when taken orally[34].
There are several drugs under investigation which inhibit some of the channel proteins. M2 forms pH-activated proton channels that are essential for normal replication of influenza A. Amantadine, the first drug against influenza A, blocks the M2 channel, impairing channel function and stopping virus replication. It was found that in response to hyperpolarizing membrane voltages, an inward whole-cell current can be blocked by amantadine and its analog, rimantadine [37]. In addition, when purified M2 protein was incorporated into the planar lipid bilayer, ionic conductance was blocked by amantadine [43]. Recently Sarah D. Cady et al. have used solid-state NMR spectroscopy and found that two amantadine binding sites exist in the M2 channel in the phospholipid bilayers. The high affinity site, located in the N-terminal channel lumen, is occupied by a single amantadine [73]. The low affinity site is on the C-terminal protein surface [73]and only high concentrations in the bilayer can reach this domain. These results indicate that there is a physical occluded mechanism in the M2 ion channel. This result is consistent with the recently solved crystal structure of the M2 protein. However, amantadine/rimantadine resistance is on the rise and amantadine-resistant mutant residues have been found in locations surrounding the high affinity site (Fig. 2) [73].
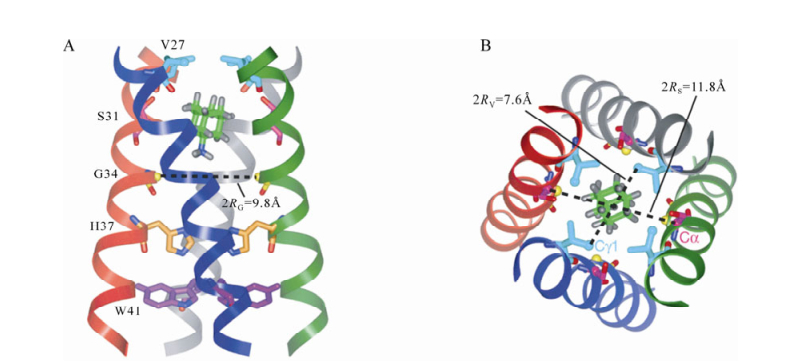
Figure 2. SSNMR structure of M2 in the lipid libayers. Amantadine (shown in both figure a and b) is in high affinity luminal site. A: Side view of the M2 channel is showing Ser 31, Val 27, Gly 34, His 37, Trp 41. The instantaneous orientation of Amt, which is slightly tilted from the channel axis, is shown. B: Top view of the M2 channel. This picture is cited from ref 73.
HIV: Human cells possess an antiviral activity that inhibits the release of retrovirus particles and other enveloped virions, and is antagonized by the HIV-1 accessory protein, such as Vpu. This antiviral activity can be constitutively expressed or induced by interferon. We call this protein-based tethers "tetherins" (B cell stromal antigen 2, BST-2/HM1.24) [33, 54]. Tetherin is an interferon-induced host cell membrane protein that blocks the release of HIV-1 virions from the cell surface [58]. In the therapeutic strategy for treating HIV infection, it is discovered that Vpu neutralizes the effect of tetherin, that provide in sight into the molecular mechanism of Vpu facilitate viron release [54, 58]. In this occasion, tetherin is an interferon-induced host cell membrane protein that blocks the release of HIV-1 virion from the cell face [58, 91]. This finding makes the concept that abrogation of Vpu function with the consequent suppression of new viable virus release from the host cell surface is a plausible therapeutic strategy in the treatment of HIV [54]. It is feasible to increase the effective cell surface density of Vpu-free tetherin by designing trans-membrane peptide decoys that target the Vpu TM α-helix. Recently, using deductive constraints and gene expression analyses Stuart J. D. Neil and his colleague identified CD317 which is a membrane protein of previously unknown function[58], as a tetherin. HIV-1 virion release requires Vpu expression in cells where CD317 expression correlated with this process, depletion of CD317 abolished this requirement [74, 90]. Vpu also respond on amantadine and derivatives, but only to very high doses [24]. Thus, inhibition of Vpu function and consequent mobilization of tetherin's antiviral activity is a potential therapeutic strategy in HIV[54, 58].
HCV: One hallmark of HCV is its high degree of sequence variability that likely contributes to its ability to establish chronic infections. Persistent infection is associated with a variable degree of liver damage and there is no available prophylactic or therapeutic vaccine to date. Although the best available treatment is a combination of polyethylene glycol-conjugated interferon alpha and ribavirin, this treatment is expensive, poorly tolerated, its outcome is largely determined by virus genotype [65] and is ineffective in -50% of patients [17], and the treatment is associated with severe side effects. The discovery that it displayed cation selectivity channel activity in vitro led to its classification within the "viroporin" family of virus-coded ion channel proteins, which includes the influenza A virus M2 protein. In vitro, HCV p7 channel activity can be inhibited by a range of compounds, including amantadine, that can reduce cation currents by high concentrations, which also inhibits the viroporin M2 of the influenza A virus. When amantadine is used in the GST-his-P7 or GST-p7 construct, it seems effective [31, 32], but has no effect on full length p7 even at concentrations up to 10 μg/mL [79]. Although it is suggested that amantadine docked into the p7 pore where is near the His-17, it does not lead to mutation at the His-17. However, it leads controversial results in HCV clinical trials [16].
Other inhibitors have subsequently been identified, long alkyl chain iminosugar derivatives, such as N-nonyl-deoxynojirimycin (NN-DNJ), which are known to inhibit endoplasmic reticulum α glucosidase Ⅰ and Ⅱ as well as HCV p7[25, 64], and hexamethylene amiloride (HMA), rimantadine[64, 68, 80]. But it is should be noted that these compounds have hitherto only been tested against genotypeⅠp7. p7 sequences contain clusters of hydrophilic amino acids predicted to line the channel lumen, implying these might determine channel opening and drug sensitivity [30], as is the case of M2. One function of p7 may be analogous to that of M2 during particle secretion [32]. Although Stephen Griffin et al. suggested that effective p7 inhibitors were not able to reduce virus titer by more than 10-fold at the concentrations used, it is provides a proof of principle for p7 to be a potential drug target. At the same time, it highlights the need for developing more potent compounds that are active against p7 sequences from multiple strains and genotypes.
Understanding the ability of these ion-forming protein structures is of great biophysical and functional importance for this class of proteins, which accounts for more than 50% of all antiviral drug targets.

 DownLoad:
DownLoad: