











-
Hepatitis C virus (HCV) is a single stranded enveloped RNA virus that belongs to the flaviviridae family. It was first discovered in 1989 as the causative agent for non-A and non-B hepatitis [20]. However, the virus had spread unknowingly for decades through blood transfusion, unsafe injection or other blood-to-blood contacts before sensitive viral diagnosis kits were developed. As a result, a huge population had been infected before mandatory blood screening was implemented in the 1990s. Over 170 million people or 3% of the world population are chronically infected with HCV, with an additional 3 to 4 million new infections each year (WHO). It is estimated that in China alone there are 20-40 million people chronically infected, representing perhaps one of the most under-appreciated health issues. Although only 25% of new infections are symptomatic, 60%-80% of patients will develop chronic liver disease, of whom an estimated 20% will progress to cirrhosis with a 1%-4% annual risk of developing hepatocellular carcinoma. Overall, HCV is responsible for 50%-76% of all liver cancer cases and two thirds of all liver transplants in developed countries. Ultimately, 5%-7% of infected patients will die from the consequences of HCV infection.
There are 7 genotypes and over 50 subtypes of HCV based on the genetic make-up of the virus. Among them genotype 1 is the most prevalent in the US, Europe, Japan and China. Unlike HIV-1, HCV does not integrate into the host genome and theoretically can be eradicated. The goal of HCV therapy is to achieve sustained virologic response (SVR), defined as HCV RNA undetectable ( < 10 IU/mL) in plasma 6 months after the end of therapy. There has been long-term follow-up of patients > 5 years after SVR, suggesting that re-infection rarely occurs ( < 1%). In other words, patients achieving SVR are essentially "cured". Also, studies have shown that the elimination of HCV infection leads to a reduction of fibrosis and the risk of developing cirrhosis and liver disease-related death.
Significant progress has been made over the past 20 years in treating hepatitis C. In the mid-1980's interferon alpha (IFN-α) was shown to reduce the levels of serum aminotransferase (ALT) and HCV RNA. However, only 6%-15% of patients achieved SVR after 6 months of IFN-α monotherapy and 13%-25% after 12 months. The addition of an oral nucleoside analog ribavirin to IFN-α in 1998 improved the response rate to 30%-40%. The mechanism of action of ribavirin is not entirely clear. It does not have a significant antiviral effect on its own but can reduce the relapse rate of IFN-α treatment. The introduction of long-acting IFN-α in 2002 not only reduced the frequency of IFN injection from three times weekly to once per week but also significantly improved treatment response. The current standard of care (SoC) for HCV infection is pegylated interferon alpha (PEG-IFN-α) in combination with ribavirin for 48 weeks in patients with genotypes 1 and 4 virus and 24 weeks in patients with genotypes 2 and 3 virus. Unfortunately genotype 1 virus, the predominant HCV genotype in developed countries and China, is also the most difficult to treat with IFN-based therapy. In patients with genotypes 1 and 4 virus the SVR rate was 41%-52% vs. 76%-82% with genotypes 2 and 3 virus [39, 94]. Moreover, both interferon and ribavirin induce significant adverse effects, including flu-like symptoms (fever and fatigue), hematologic complications (leukopenia, thrombocytopenia), and neuropsychiatric issues (depression, insomnia) associated with interferon and significant hemolytic anemia associated with ribavirin. Also, ribavirin is teratogenic and cannot be given to pregnant women. Therefore, the majority of HCV patients are not being treated with current SoC. More effective and better tolerated therapies are therefore urgently needed, which is the subject of this review.
HTML
-
The life cycle of HCV has been well studied and has revealed many potential targets for novel therapies. HCV, an enveloped RNA virus, first enters the cells through specific interactions of viral glycoproteins (E1 and E2) with cell surface receptors CD81 [112] human scavenger receptor class B type Ⅰ (SR-B1) [131], tight junction proteins Claudin-1 [30] occludin [113], and likely other cell surface proteins. Following attachment, the HCV nucleocapsid is released in the cytoplasm as a result of a fusion process between viral and cellular membranes, which is pH-dependent and is mediated by clathrin-dependent endocytosis. Decapsidation of viral nucleocapsids releases positive-strand genomic RNA, which serves as the template for the synthesis of the HCV polyprotein in the cytoplasm. The 5'-UTR of HCV contains an internal ribosomal entry site (IRES), which mediates cap-independent initiation of HCV polyprotein translation by recruiting cellular proteins eukaryotic initiation factors eIF-2 and 3. The 9.6 kb HCV RNA genome encodes a single large open reading frame corresponding to a polyprotein precursor of about 3, 000 amino acids, which is proteolytically cleaved into ten individual proteins, in the order of C-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B (Fig. 1). Host signal peptidase and signal peptide peptidase are responsible for the cleavage at the junctions of core-E1, E1-E2, E2-p7 and p7-NS2. The zinc-dependent NS2-3 autoprotease ensures cis-cleavage of NS3 from NS2. The NS3 serine protease, together with its cofactor NS4A, catalyzes cis-cleavage at the NS3-NS4A junction and trans-cleavage at all downstream junctions including NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B. The viral non-structural proteins then form the replication complex with cellular components and nascent RNA strands on an ER membrane derived structure named mem-branous web. The positive-strand genome RNA serves as the template for the synthesis of a negativestrand RNA, which in turn serves as the template to produce 5 to 10-fold excess positive-strand RNA that will be used for polyprotein translation or packaging into new virus particles. The virions are assembled on lipid droplets (LD), which are located at endoplasmic reticulum (ER)-derived bilayer membranes. The Core protein of HCV localizes on the monolayer membrane that surrounds the LD. It recruits non-structural (NS) proteins to the LD-associated membrane. E2 also localizes around the LD. The positive strand HCV RNA genome is encapsidated with the structural proteins. The viral particle is probably enveloped through budding into the ER lumen and then trans-ported through Golgi to be released.
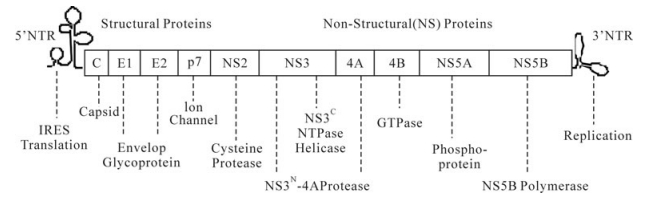
Figure 1. HCV genome.
As shown in Fig. 2, every step of the HCV life cycle could potentially be intervened with antiviral agents. All ten HCV proteins have been pursued as antiviral targets. Among them, drug discovery efforts have been mainly focusing on the NS3-4A serine protease and the NS5B RNA-dependent RNA poly-merase, both of which have enzymatic activities essential for viral replication and are considered highly druggable targets partly because of the success of antiretroviral therapy targeting HIV-1 protease and polymerase. In addition, cellular proteins are involved in every step of the viral life cycle and can also be considered as potential antiviral targets. Host factors not only provide a complementary antiviral strategy but also may have the advantage of creating higher genetic barriers to resistance. Cyclophilins, a family of cellular PPIase required for viral replication, represent such a strategy. A number of other potential host targets have been identified through siRNA screens [9, 77, 104, 145, 153].
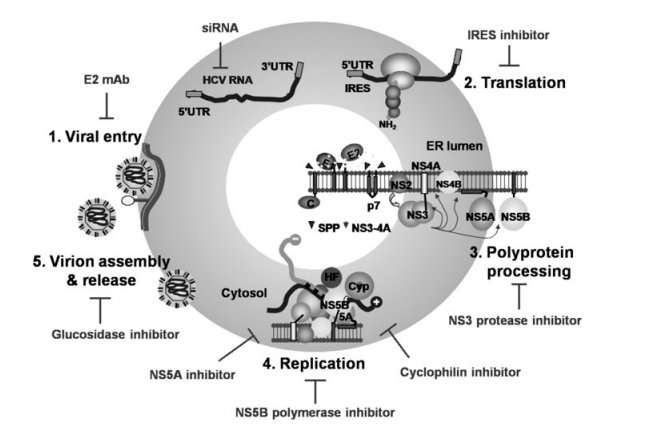
Figure 2. HCV life cycle and antiviral targets. (HF=host factor, Cyp=cyclophilin)
Drug discovery effort on HCV has long been hampered by the lack of an in vitro virus culture system and suitable animal models. The establishment of a subgenomic HCV replicon system in 2000 greatly facilitated basic research studying viral replication in vitro as well as HCV drug discovery efforts. The subgenomic replicon contains all the non-structural proteins of HCV that are required for autonomous replication of viral RNA in a human hepatoma cell line, Huh-7 [8, 84]. However, it lacks viral structural proteins and therefore does not produce infectious virus. Another in vitro culture system, HCV pseu-dotyped viral particle (HCVpp), generated from lentivirus replacing native glycoproteins with HCV E1 and E2, provides a useful tool to study viral entry [5, 58]. However, only the newly discovered genotype 2a HCV (JFH-1 strain) recapitulates the complete viral life cycle [80, 146, 159]. A genotype 1 virus (H77 strain) can also infect Huh-7 cells in vitro, albeit at much lower infectivity compared to that of the JFH-1 strain [156]. Numerous attempts have been made to culture HCV isolated from patient serum using primary human hepatocytes, however no robust, reproducible method has been established to date. The development of animal models has been equally challenging. Chimpanzees are the only immunocompetent animals that can be chronically infected with HCV, but their use is restricted by ethical concerns, limited availability and prohibitively high cost [14]. SCID mice with human hepatocytes repopulated in the mouse liver can be infected with HCV and provide a useful tool for compound efficacy testing and possibly PK and toxicology studies [98]. However, these mice are also of limited availability and substantial variability and cannot be used to study pathology or immunology aspects of infection. Despite these constraints, more than two dozen novel HCV inhibitors have progressed beyond preclinical development and demonstrated clinical efficacy in HCV patients. Key classes of HCV inhibitors in development are listed in Table 1, some of which are discussed in detail below.

Table 1. Key classes of HCV inhibitors in development. (NI=nucleoside inhibitor, NNI=non-nucleoside inhibitor)
-
The NS3 protein of HCV has dual functions: the N-terminal one third of the protein contains a chymotrypsin-like serine protease domain whereas the C-terminal portion of the protein is a helicase/NTPase. Together with the NS4A cofactor the NS3 protease is responsible for proteolytic cleavage of the HCV polyprotein at four junctions, NS3-4A (cis or self-cleavage), 4A-4B, 4B-5A and 5A-5B, and thus is essential for viral replication [4, 45, 143]. The substrate specificity of NS3 protease has been well characterized. The cleavage sites recognized by the NS3-NS4A protease have the following sequence in common: Asp/GluXXXXCys/Thr-Ser/Ala, with trans-cleavages occurring downstream of a cysteine residue and the cis-cleavage occurring downstream of a threonine residue. Both HPLC and FRET-based NS3 protease assays have been developed, which measure the cleavage of a peptide substrate by either the protease domain or the full-length NS3 protein coupled with an NS4A peptide [70, 140]. The assays are quite amenable for high-throughput screening of small molecule inhibitors. However, none of the screening efforts led to any promising leads for NS3 inhibitors. Thus, the discovery of specific inhibitors of NS3 protease has been largely depending on structure-based drug design.
The crystal structure of the NS3 protease was first solved in 1996 [64, 86], which revealed a shallow, featureless substrate binding pocket, suggesting that the design of small inhibitors could be challenging. The initial peptidomimetic inhibitors were based on the decapeptide (P6-P4') natural substrate of NS3, which was the minimum required length of peptide substrate for efficient cleavage. Using non-cleavable active-site analogs, the substrate-based inhibitor can be further truncated to P4-P1', a more drug-like dimension [70]. In parallel, it was discovered that the products of the natural substrates cleavage were themselves inhibitors of NS3 protease and therefore can also be used as the starting point for inhibitor design [83, 137]. Initial combinatorial screen of the natural cleavage product sequences led to a series of very potent hexapeptide inhibitors. Over the past 15 years, tremendous effort has gone into optimizing these peptidomimetic inhibitors through extensive substitutions at every position of the molecule, which have greatly improved the potency and drug-like properties of the inhibitors. Now there are at least a dozen NS3 protease inhibitors in development, which can be generally divided into two groups based on differences in chemical structure as well as mechanism of inhibition. One group comprises substrate based inhibitors including telaprevir (VX-950) and boceprevir (SCH 503034), which contain an electrophile warhead (instead of the scissile bond) that engages the catalytic serine of NS3 active site in a covalent but reversible interaction, so called "serine trap". The others are non-covalent product based inhibitors which are either carboxylic acid such as BILN2061 or have an acylsulfonamide at P1' like ITMN-191.
BILN2061 (ciluprevir) was the first NS3 protease inhibitor demonstrating clinical efficacy. Treatment with 200 mg bid (twice daily) of the compound in genotype 1 HCV patients resulted in a rapid viral load reduction of 3 log10 copies/mL after only 2 days [53, 69]. Unfortunately the development of the compound was terminated due to cardiotoxicity findings in animal studies [120].
Currently, the most advanced protease inhibitors in development are telaprevir and boceprevir in Phase Ⅲ clinical trials. Telaprevir is a ketoamide inhibitor of NS3 protease with moderate in vitro potency (NS3_Ki= 44 nmol/L, HCV replicon EC50=354 nmol/L) [78, 110]. In a Phase Ib trial, telaprevir monotherapy for 14 days reduced HCV viral load by 3.5 to 4.8 log10, with the optimal dosing determined to be 750 mg every 8 h (q8h) because of the relatively short half-life of the compound [119]. Subsequent Phase Ⅱ trials in treatment naive patients showed that the combination of telaprevir with PEG-IFN-α and ribavirin increased sustained virologic response (SVR) by about 20% compared to current standard of care [52, 97]. However, adverse events (AEs) were more common in the telaprevir treatment group, including skin rash, gastrointestinal events and anemia, which resulted in higher discontinuation rates compared to PEG-IFN-α/ ribavirin alone. In patients previously failed PEG-IFN-α/ribavirin therapy, the triple combination also demonstrated significant improvement 51%-52% SVR vs. 14% with SoC [93]. Three large Phase Ⅲ trials are currently on-going. New Drug Application (NDA) filing is expected in 2011. Boceprevir is another α-ketoamide inhibitor of similar potency (replicon EC50=200 nmol/L) [90]. In genotype 1 HCV patients previously not responding to IFN and ribavirin therapy, 400 mg q8h of boceprevir monotherapy resulted in a 1.61 log10 viral load reduction. Combination of boceprevir with PEG-IFN-α2b resulted in a 2.88 log10 viral load reduction compared to only 1.3 log10 with PEG-IFN-α2b alone [129]. In a Phase Ⅱ trial with treatment naive patients, triple therapy including 800 mg q8h boceprevir resulted in 67%-75% SVR compared to 38% with PE-IFN-α2b/ ribavirin. However, treatment with boceprevir caused higher incidence of anemia in addition to fatigue, nausea and headache that were typically associated with SoC [67]. Phase Ⅲ trials with boceprevir in treatment naive and failure patients are also on-going.
As shown in Table 1, a number of other NS3 protease inhibitors are in early phases of clinical development. Many of these compounds have shown much improved potency (replicon EC50 < 10 nmol/L) and longer half-life which enables more convenient dosing (once or twice daily). The efficacy and safety of these potentially better second generation protease inhibitors are being evaluated in further clinical trials.
-
The NS5B protein of HCV is an RNA-dependent RNA polymerase. It is responsible for synthesis of both positive and negative strands of HCV RNA and thus is essential for viral replication. NS5B is anchored to the ER membrane through its C-terminal 21 amino acids and catalyzes the polymerase reaction on the cytosolic side of ER. There is no mammalian homolog of NS5B in terms of subcellular localization and template specificity, suggesting it may be possible to identify selective inhibitors. NS5B that lacks the C-terminal membrane-anchoring domain can be efficiently expressed in E. coli, which allows the establishment of a robust polymerase assay in vitro measuring the incorporation of ribonucleoside triphosphate to homopolymer or heteropolymer RNA template. In contrast to NS3 protease, high-throughput screening of small molecule libraries successfully led to the identification of multiple classes of NS5B inhibitors. The structure of NS5B was solved in 1999 [1, 13, 76], which helped characterizing binding of these inhibitors to NS5B. The structure of NS5B, like many other viral polymerases, resembles the shape of a right hand consisting of finger, thumb and palm domains. There are two major classes of polymerase inhibitors, nucleosides and non-nucleosides.
Nucleoside inhibitors (NIs) bind competitively with natural nucleoside triphosphate substrates to the active site of polymerase and, once incorporated, serve as chain terminator to block further extension of viral RNA nascent strand. The first nucleoside analog demonstrating clinic efficacy against HCV is valopicitabine (NM283) [111]. At 800 mg/day it reduced viral load by 1.2 log10 after 15 days of treatment. However, further development of the compound was terminated due to dose-limiting GI toxicity and insufficient efficacy/safety benefits. The second HCV NI, R1626, achieved a more profound viral load reduction of 3.5 log10 after 14 days at the highest dose tested, 4, 500 mg twice per day (bid). Combination of 1500 mg bid R1626 with PEG-IFN-α2a/ribavirin resulted in a 5.2 log10 viral load reduction after 4 weeks [114, 122]. However, further development of R1626 was also discontinued due to higher incidence of neutropenia. Currently, the most advanced HCV NI is R7148. In the Phase Ⅰ monotherapy trial, 1, 500 mg bid of R7148 resulted in a 2.7 log10 viral load reduction after 14 days. Combination of 1000 mg or 1500 mg bid of R7148 with PEG-IFN-α2a/ribavirin resulted in 85% rapid virological response (RVR) compared to 19% with SoC [124]. Two liver-targeting prodrugs of nucleoside analogs, IDX184 and PSI-7851, recently entered clinical development. These compounds are designed to achieve higher concenrations of the active metabolites in the liver while reducing systemic exposure thereby limiting potential side effects. IDX184, a nucleotide prodrug of 2'-methyl guanosine, resulted in a 0.47 log10 viral load reduction after dosing 25 mg once daily (qd) for three days and 0.74 log10 at 100 mg qd. Combination of 50 mg qd IDX-184 and PEG-IFN-α/ribavirin resulted in a 3.66 log10 reduction after 14 days vs. only 1.7 log10 with PEG-IFN-α/ribavirin alone [68]. PSI-7851 is a phos-phoramidate prodrug of β-D-2'-deoxy-2'-fluoro-2'-C-methyluridine-5'-monophosphate. When administered as a monotherapy, 400 mg qd of the compound suppressed HCV RNA by 1.95 log10 after 3 days[125]. Nucleoside inhibitors are generally less potent than other classes of HCV inhibitors. However, it appears to be more difficult to develop resistance against nucleoside inhibitors vs. non-nucleoside polymerase inhibitor or protease inhibitor[96]. This is mainly due to the fact that nucleoside inhibitors bind to the highly conserved active site of NS5B. Any mutation at the active site conferring resistance also leads to a significant cost to the fitness viral polymerase and replication.
Non-nucleoside inhibitors (NNI) of NS5B are non-competitive with regard to nucleoside substrate and bind to the surface of the protein. Interestingly, there have been at least four allosteric binding pockets identified for HCV NNIs.
NNI site 1 is also referred as thumb pocket 1. Inhibitors of this site are hypothesized to displace the Λ finger loop from the upper thumb domain of NS5B and interfere with a conformational change required during RNA synthesis. BILB1941 was the first site 1 NNI that reported proof-of-concept (PoC) clinical efficacy [29]. It has a replicon EC50 of 153 nmol/L and 84 nmol/L against genotype 1a and 1b, respectively. A greater than 1 log10 viral load reduction was achieved when patients received 450 mg q8h of the compound for 5 days. Unfortunately further development of the compound was terminated due to GI intolerance. Recently it was reported that a follow-up compound, BI-207127 achieved a dose-dependent viral load reduction in the range of 0.6-3.1 log10 after 5 days [71]. Another site 1 compound, MK-3281, an indole-based inhibitor of 40 nmol/L replicon EC50, also reported clinical efficacy, with much greater viral load reduction in genotype 1b patients (3.75 log10) than genotype 1a patients (1.28 log10) after dosing 800 mg bid for 7 days [12].
NNI site 2, also known as thumb pocket Ⅱ, is a hydrophobic pocket located at the base of the thumb domain. A series of thiophene carboxylic acid based inhibitors were discovered to bind to this pocket. The first compound of this series demonstrating clinical efficacy was VCH-759, which had an EC50 of 0.34 and 0.27 µmol/L against genotype 1a and 1b replicon, respectively. Ten days of monotherapy with VCH-759 at 400 and 800 mg tid (three times daily) resulted in 1.9 and 2.5 log10 viral load reduction in HCV patients [25]. A second, slightly more potent compound, VCH-916 (1a/1b replicon EC50=79/110 nmol/L), produced a 1.5 log10 viral load reduction after 3 days [73]. Currently the most advanced inhibitor of the series is VCH-222 (VX-222), which has an EC50 of 65 and 41 nmol/L against genotype 1a and 1b replicon, respectively. In genotype 1 HCV patients, 250-750 mg bid or 1500 mg qd of VX-222 resulted in a > 3 log10 viral load reduction after 3 days [126]. Another class of site 2 inhibitors is represented by filibuvir (PF-868554), which is partially cross-resistant with thiophene carboxylic acids as their binding pockets overlap. The compound had a replicon EC50 of 59 nmol/L in vitro [135]. Monotherapy with the compound at 450 mg bid or 300 mg tid resulted in a maximum viral load reduction of 2.1 log10. These site 2 inhibitors all lost activity against other HCV genotypes, presumably due to sequence variation (polymorphism) around the binding pocket.
NNI site 3 is located at the palm domain of NS5B. Benzothiadiazine inhibitors targeting site 3 were first discovered in 2001 and subsequently followed up by several groups [18, 51, 105, 117, 133]. ANA598 belongs to this series and has great in vitro potency, particularly against genotype 1b (replicon 1b EC50=2.8 nmol/L, 1a EC50=29.2 nmol/L). In a three-day Phase Ⅰ study, a median viral load reduction of 2.3-2.9 log10 was achieved with 200-800 mg bid of the compound. The antiviral effect was more pronounced in genotype 1b patients than in 1a patients, consistent with in vitro potency of the compound [74]. Combination of 200 mg bid ANA598 and SoC for 12 weeks reduced viral load to undetectable level in 73% of patients. However, a higher incidence of skin rash was observed with ANA598 treatment. Further evaluation of the com-pound is on-going.
NNI site 4, also known as palm site Ⅱ, partially overlaps with site 3 but is closer to the active site and the junction between the palm and thumb domains. A series of benzofuran based inhibitors were identified to bind to site 4. The most notable compound is HCV-796, a very potent inhibitor of both genotype 1a and 1b HCV (replicon EC50=10 nmol/L) [57]. In a Phase 1b trial patients receiving 500-1000 mg bid HCV-796 monotherapy had a peak viral load reduction of 1.4 log10. However, the viral load started rebounding in most patients at day 4, which was associated with the emergence of resistance mutations. In a Phase Ⅱ trial, combination with PEG-IFN-α2b resulted in 3.3-3.5 log10 HCV RNA reduction at day 14 vs. only 1.6 log10 with PEG-IFN-α2b alone. However, the compound was discontinued due to significant hepatotoxicity findings in two patients receiving the compound in combination with PEG-IFN-α2b/ribavirin for 12 weeks [31].
-
Apart from NS3 and NS5B, the other viral proteins have also been pursued as potential antiviral targets. The most promising one is NS5A. NS5A is a multifunctional protein. NS5A interacts with NS5B and is part of the replication complex, thus it is required for viral replication. More recently it was demonstrated that NS5A is also involved in viral assembly. NS5A exists in both basally and hyperphsophorylated forms, the function of which may be regulated by cellular kinases. NS5A protein is consisted of three domains. The structure of domain 1 was solved recently by two independent groups [85, 141], interestingly revealing two different conformations, which were hypothesized to be associated with different roles that NS5A may play at different steps of viral life cycle.
Besides the intriguing biology associated with NS5A, the discovery of a NS5A inhibitor BMS-790052 has greatly increased the interest on this protein as a drug target. It is the most potent HCV inhibitor reported to date, with a replicon EC50 of 9 pmol/L against genotype 1b and 50 pmol/L against genotype 1a. The exceptional potency of the compound also translated to clinical efficacy: a single dose of BMS-790052 resulted in a 3.6 log viral load reduction in HCV patients after 48 h which was maintained for 6 days [41]. The compound is currently being evaluated in Phase Ⅱ trials in combination with PEG-IFNα/ribavirin. Several other NS5A inhibitors are also reportedly in early clinical or preclinical development (Table 1).
It should be noted that the mechanism of action of these compounds has not been completely elucidated. They were claimed to be NS5A inhibitors mainly because they select for specific resistant mutations in NS5A. Some of the compounds were shown to modulate the phosphoylation of NS5A [75]. However, to date no data has been published demonstrating a direct binding of the inhibitors to NS5A protein. Besides the cell-based replicon assay, there is no defined function of NS5A that can be used to measure and optimize the activity of the inhibitors.
NS4A is the co-factor of the NS3 protease. A series of acyl thiourea inhibitors were identified through replicon screening. It was proposed that these compounds bind to NS4A and interfere with the interaction between NS3 and NS4A. The lead compound ACH 806 (GS-9132) reduced viral load by 0.91 log10 at 300 mg bid for 5 days in HCV patients but was terminated due to nephrotoxicity [85, 141, 155].
NS4B is responsible for anchoring the replication complex to the ER membrane and is required for viral replication. Recently it was reported that NS4B also contributes to virus assembly and release. There have been several reports of potential NS4B inhibitors [19, 27, 121, 134], mainly based on the observation that these inhibitors select for specific resistance mutations in NS4B. Because of the lack of a functional assay for NS4B and the absence of direct-binding data, the mechanism of action of these inhibitors remains to be elucidated.
The p7 protein of HCV was shown to be required for viral replication in chimpanzees [128]. The function of p7 has been unknown until recently several groups showed that p7 has cation channel activity in vitro [47, 109] and appears to play an important role during virion secretion in culture [60, 138]. The structure of p7 was solved recently [87], displaying some similarity to M2 ion channel of influenza virus, both of which can be blocked by the inhibitor amantidine [47]. P7 can also be inhibited by iminosugars [109] and hexamethylene amiloride [115]. Compounds that block its activity in vitro also inhibit viral particle production in cell culture [46, 139]. BIT225, a compound with known activity against HIV-1 Vpu ion channel [63], blocks p7 and is currently in early clinical trials for HCV [88].
The C-terminal two-thirds of NS3 is a helicase/ NTPase, which has been well characterized. The structure of the NS3 helicase is also available. However, helicase is traditionally a difficult target. Despite a number of screening and early drug discovery efforts [10, 11, 44, 136, 144], no potent and selective NS3 helicase inhibitors have been identified that are suitable for further development.
Cleavage of HCV polyprotein between the NS2 and NS3 is mediated by an autoproteolytic activity that requires both the C-terminal portion of NS2 and the N-terminal of NS3. Assays have been established to measure the protease activity, which could enable screen for inhibitors [26, 142, 150]. However, NS2 remains to be a difficult target because of the hydrophobic nature of the protein and the challenge to inhibit an autoproteolytic reaction. It was recently discovered that NS2 is also involved in viral assembly and production, however the autoprotease activity of NS2 does not appear to be required [59].
-
There are only ten viral proteins of HCV and not all of them are druggable targets, but there many more host proteins involved in viral replication, which greatly expand the list of potential antiviral targets. Targeting host factors also has the advantage of presenting a higher genetic barrier to resistance. One of such targets is cyclophilin. Cyclophilins are a family of highly conserved cellular peptidyl-prolyl cis-trans isomerases (PPIase), which are involved in many cellular processes such as protein folding and trafficking. It has been shown that cyclophilins particularly cyclophilin A (Cyp A) is required for HCV replication. Knock-down of Cyp A with specific siRNAs blocked HCV replication [40, 99, 100, 147]. The HCV inhibitory activity of cyclosporin analogs correlated with their cyclophilin-binding affinity, but not immunosuppressive or P-gp inhibitory activity [89]. Although the functions of cyclophilins in HCV remain to be fully elucidated, increasing evidence suggests that cyclophilins (mainly A and B) are involved in HCV replication by (1) interacting directly with viral proteins (NS5A and NS5B) as part of the replication complex and/or (2) mediating the correct folding and trafficking of viral proteins to the site of replication (cytosolic side of ER membrane) through their PPIase activity [15-17, 21, 32, 40, 43, 49, 50, 62, 81, 123, 148, 154]. Cyclophilin inhibitors block the interaction of cyclophilins with HCV proteins and hence the formation of a functional viral replication complex.
Three cyclophilin inhibitors have entered the clinic and shown efficacy in HCV patients. NIM811, a non-immunosuppressive cyclosporin analog, is a potent HCV inhibitor in vitro [89]. The combination of NIM811 with IFN-α and NS3 protease or polymerase inhibitors not only enhanced anti-HCV activity but also helped to suppress the emergence of resistance [95]. In a Phase 1b trial in genotype 1 HCV patients who had relapsed in prior interferon therapy, patients receiving 600 mg bid NIM811 plus PEG-IFN-α2a had an HCV viral load reduction of 2.78 log10 compared to only 0.58 log10 with PEG-IFNα-2a alone [72]. Alisporivir (Debio-025) is a more potent cyclosporin analog [106]. In a Phase Ⅰ trial in HIV-HCV co-infected patients, 1200 mg bid of alisporivir monotherapy resulted in a 3.4 log10 reduction of HCV RNA after 14 days [37]. In a Phase Ⅱ combination study, 600 mg qd alisporivir plus PEG-IFN-α2a led to 4.6 log10 viral load reduction after 28 days in genotypes 1 and 4 patients and 5.9 log10 in genotype 3 patients [35]. The third cyclosporin analog that has shown clinical efficacy is SCY-635, which had a 2.2 log10 viral load reduction in a 15-day monotherapy trial in genotype 1 HCV patients [55].
Alpha-glucosidase Ⅰ is another host target being pursed, which is involved in glycoprotein processing and is important for viral maturation and release. Inhibition of alpha glucosidase leads to misfolding of HCV envelop protein thus blocks viral assembly and release [151]. Monotherapy with an alpha-glucosidase Ⅰ inhibitor, celgosivir, resulted in only a modest antiviral effect. In a 12-week Phase Ⅱ trial, only 2 out of 35 patients had greater than 1 log10 viral load reduction [157]. Combination of 400 mg celgosivir with PEG-IFNα/ribavirin resulted in greater than 2 log10 viral load reduction in 45% patients vs. 10% with PEG-IFN-α/ribavirin only [61]. Further combination trials with different dosing regime are currently on-going.
Nitazoxanide is a drug previously approved for parasitic infestations. Recently, it was shown to inhibit HCV through inducing phosphorylation of eukaryotic initiation factor 2α, a known mediator of host antiviral defense [28, 66]. In a Phase Ⅱ study in genotype 4 patients in Egypt, the combination of nitazoxanide with PEG-IFN-α2a and ribavirin resulted in 79% SVR vs. 50% with PEG-IFN-α2a and ribavirin alone [127].
-
Several new forms of long-acting IFNs are in clinical development. The most advanced is albinterferon-α2b (albIFN), a fusion protein of human albumin and IFN-α2b. It has an extended half-life of 144 h, which is even longer than those of pegylated IFNs and allows dosing every two or four weeks while maintaining a comparable efficacy and safety profile [3, 101, 158].
Toll-like receptor TLR7 recognizes single-stranded RNA virus and activates type 1 IFNs as part of innate immune response. Small molecule agonists of TLRs such as isatoribine could exert antiviral effect through activation of TLR and IFN pathway. Isatoribine and an oral prodrug of isatoribine, ANA975, showed clinical efficacy in HCV patients [56, 152]. However, further development of these drugs was discontinued due to significant side effects and insufficient therapeutic window. In 2009, it was reported that a novel oral TLR7 agonist ANA773 demonstrated significant antiviral response in hepatitis C patients. Patients receiving 2000 mg of ANA773 every other day for 10 days has a mean maximal viral load decline of 1.3 log10, compared to 0.3 log10 with placebo. No serious adverse events were reported. Further clinical trials of ANA773 in combination with other HCV agents are expected.
There have also been limited efforts in developing therapeutic vaccines for HCV. GI-5005 is a heat-inactivated recombinant Saccharomyces cerevisiae that has been genetically modified to express HCV NS3 and core proteins [48]. In a Phase Ⅰ trial, GI-5005 showed a modes antiviral effect reducing viral load by 1.4 log10. It is currently being evaluated in triple combination therapy with SoC. A number of other vaccine strategies such as DNA and T cell based vaccines have also been pursued. IC41, a peptide-based vaccine is currently in early clinical deve-lopment [33, 34, 65, 149].
-
The first generation of orally active direct acting antiviral agents (DAA), specifically NS3 protease inhibitors, are expected to be approved within two years to be used in combination with current SoC (PEG-IFN-α and ribavirin). These new triple combination therapies will significantly improve treatment response over current SoC. However, the side effects associated with these new oral agents in addition to the significant side effects already caused by IFN and ribavirin suggest that even less patients can tolerate the new therapies. Moreover, the first generation DAA has short half-life and requires frequent dosing (q8h). Patient compliance could become a real issue which may significantly compromise the efficacy and utility of the drugs.
With multiple classes of small molecule DAAs in development, the future objective of HCV therapy is an IFN-free, oral cocktails of DAAs, similar to the highly active antiretroviral therapy (HARRT) for HIV-1 infection. The first of such combinations has yielded promising results in the clinical trial. The combination of an NS3 protease inhibitor RG7227/ ITMN-191 and an NS5B polymerase inhibitor RG7128 administered without PEG-IFN-α or ribavirin for 13 days reduced HCV RNA to an undetectable level in 88% of treatment-naive patients and 50% of null responders to prior IFN therapy [42]. Several other clinical trials exploring different oral combinations have already been initiated, including telaprevir plus VX-222, BMS-790052 plus BMS-650032, and GS-9256 plus GS-9190 [22].
Despite these progresses, it remains to be determined whether an IFN-free regimen is capable of completely clear the virus, i.e. cure HCV infection. The main issue for DAAs is the development of resistance. It has been demonstrated both in vitro and in patients that drug-resistant mutants can emerge quickly, even with the most potent inhibitors of viral protease or polymerase [130]. This has been attributed to several factors: (ⅰ) HCV replicates at a high rate in patients, producing an estimate of 10[10-12] virions per day [103]; (ⅱ) the RNA-dependent RNA polymerase of the virus lacks proof-reading function and has an error rate of about 10-4 mutations per genome per replication cycle. As a result there is an extremely high degree of heterogeneity of viral population (quasispecies) in each patient; (ⅱ) since viral targeted inhibitors typically bind to a defined pocket of a viral protein, typically a single mutation in viral genome is sufficient to disrupt the binding of inhibitor and lead to resistance. Theoretically all the possible single, double or even triple mutations are already pre-existing in HCV patients, therefore it was estimated at least three DAAs are required to completely suppress the emergence of resistance. Compounds with relatively high resistance barrier such as cyclophilin inhibitors could provide the key advantage in an IFN-free regimen. Thus, the combination of host and viral targeted inhibitors could be an attractive strategy in maximizing antiviral efficacy and suppressing the emergence of resistance.

 DownLoad:
DownLoad: