











-
Cervical cancer is the second leading cause of cancer death among women worldwide, and more than 95% of cervical cancers contain human papillomavirus (HPV), particularly the high-risk HPV type 16 (HPV16)[9]. Though HPV screening can increase the likelihood of detecting premalignant lesions, it cannot eliminate HPV16 infection and still is difficult to adopt in developing countries. Additionally. there is still no effective countermeasures in clinical trials of cervical cancer in mid and late stage. Therefore the development of therapeutic vaccines targeted to HPV16 of vital importance in cervical cancer treatment.
Two HPV early oncoproteins, E6 and E7, are selectively retained and expressed in cervical tumors and are responsible for the malignant transformation[2, 5, 8]. These oncogenic proteins therefore represent ideal target antigens for developing vaccines and immunotherapeutic strategies against HPV-associated cervical cancer and its precancerous lesions. Recently, studies have clearly shown that a recombinant E6/E7 fusion protein is significantly more efficient in inducing antitumor protection than immunization with the E6 or E7 oncoprotein alone [12].
However, E6 and E7-based vaccines have some potential risks because of the oncogenic property of E6 and E7 and so appropriate genetic mutations to HPV16 E6 and E7 are indispensable to ensure the safety of the vaccine. It is reported that the mutants of L57G, C113R of the E6 gene and C24G, E26G of the E7 gene can not only eliminate their transformation capability but also retain the very good stability and antigenicity of E6 and E7 proteins[3, 6, 10].
In this research, a recombinant HPV16 E6E7 fusion gene with the codon-modification for the human gene was constructed, and site-directed mutagenesis at both L57G, C113R in the E6 protein and at C24G and E26G in the E7 protein were introduced into HPV16 E6E7 fusion protein [patent pending (CN 101100672)]. The optimized and recombinant HPV16 E6E7 mutant not only lost its transformation capability into NIH 3T3 cells and tumorigenicity in SCID mice, but also maintains good stability and antigenicity. Hopefully, the recombinant HPV16 E6E7 gene was constructed in this work can undergo further research as a potential therapeutic vaccines for HPV16-associated tumors.
HTML
-
Plasmid DNA construction
The HPV16 E6E7 fusion gene with the termination codon of E6 gene removed was optimized on the basis of the codon usage for mammalian cell expression.The optimized HPV16 E6E7 fusion gene (HPV16 ofE6E7) was then synthesized with the Splicing by Overlapping Extension PCR (SOE-PCR) (data not shown here). The HPV16 ofE6E7 gene with Kpn Ⅰ and Xba Ⅰ sites was cloned into the pUC18 vector that was cut using Kpn Ⅰ / Xba Ⅰ to generate pUC18/HPV16 ofE6E7.
For the generation of the pcDNA3.1(+)/ HPV16 omf E6E7 plasmid, a mutant of the HPV16 ofE6E7 gene at L57G, and C113R in the E6 protein and at C24G and E26G in the E7 protein with BamH Ⅰ/EcoR Ⅰ sites was amplified from pUC18/HPV16 ofE6E7 plasmid by SOE-PCR using the primers listed in Table 1.

Table 1. Primers used in this study
For generation of pcDNA3.1(+)/ HPV16 of E6E7 plasmid, HPV16 of E6E7 gene with BamH Ⅰ / EcoR Ⅰ sites was amplified from pUC18/ HPV16 ofE6E7 plasmid by PCR using primers E6Ubam and E7Leco (Table 1).
Both PCR products were then digested with BamH Ⅰ/ EcoR Ⅰ enzymes and cloned into the pcDNA3.1(+) vector that was cut by BamH Ⅰ / EcoR Ⅰ. All plasmid constructions were verified by DNA sequencing.
Cell culture and transfections
The NIH 3T3 cell line was purchased from American Type Culture Collection (Rockville, MD). TC-1 cells expressing HPV16 E6 and E7 were kindly provided by Dr. T.C. Wu of Johns Hopkins University. Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 1 mmol/L L-glutamine, 100 U/mL penicillin and 100 mg/mL streptomycin. For stable transfection, pcDNA3.1 (+)/ HPV16 of E6E7 and pcDNA3.1(+)/ HPV16 omf E6E7 were transfected into NIH3T3 cells using a Lipofectamine 2000 liposome transfection kit (In-vitrogen, USA). The pcDNA3.1 empty vector transfection group and the blank control group (only liposome was added, and there was no vector DNA transfected) were established. Putative transfectants were then selected by antibiotic resistance in cell culture medium containing 800 μg/mL G418. After 3 weeks of culture in the presence of G418, the remaining cells were isolated with cloning cylinders and transferred into 24-well dishes.
Immunofluorescence imaging
To identify cell clones that carried the transfected HPV16 ofE6E7 and HPV16 omfE6E7 cDNAs, cells selected with G418 and growing in multi-tissue culture dishes (Corning, USA) were rinsed once with PBS, fixed for 30 min in iced 4% paraformaldehyde, washed in PBS, and permeabilized with 1% Triton X-100 in PBS. Cells were then treated with monoclonal antibody of mouse anti-HPV16 E7 (Santa Cruz, USA) for 1 h, washed 3 times in PBS, and incubated with Goat Anti-mouse IgG-FITC Antibody (Santa Cruz, USA). After extensive washing in PBS, the cells were viewed with a fluorescence microscope.
Western Blotting assay
NIH 3T3 cells (106) that had been transfected with pcDNA3.1(+)/ HPV16 ofE6E7, pcDNA3.1(+)/ HPV16 omfE6E7 and pcDNA3.1 empty vector were digested with buffer which contained 62.5 mmol/L Tris-HCl, pH 6.8, 2% SDS, 5% 2-mercaptoethanol, 10% glycerol. The cell lysis samples were spun at 13 000 r/min for 5 min, the supernatants were then collected and loaded onto the 10% SDS-poly-acrylamide gel electrophoresis (SDS-PAGE). Protein on the gels was electro-transferred onto nitrocellulose membranes, and blocked with blocking buffer containing 5% of non-fat milk, the membrane was then incubated with primary antibodies against HPV16 E6 (polyclonal antibody of goat anti-HPV16 E6, Santa Cruz) at room temperature for two hours. After washing three times in PBS, 10 min each wash, the membrane was incubated with anti-goat Ig horseradish peroxidase linked secondary antibody (GE Healthcare Bio-Science, USA) at room temperature for one hour, and washed three times in PBS, 10 min each wash. The protein bands were then developed using an enhanced chemiluminescence (ECL) kit according to the manufacturer's specifi-cations (GE Healthcare Bio-Sciences), and visualized using the Gene-Genius Imaging System (Syngene, Frederick, MD, USA). The densities of the Western Blot bands were evaluated using the ImageJ Image Analysis Software.
Indentification of transforming activity
Colony formation assay
Colony formation assay was determined as described previously[1], with some modifications. Briefly, the NIH3T3 cell lines (1×103) expressing HPV16 ofE6E7 or HPV16 omfE6E7 were suspended in DMEM medium containing 10% FCS and 0.3% agarose and subsequently overlayed onto a solidified layer of DMED medium containing 10% FCS and 0.5% agarose. Cells were cultured for 21 days and cellular foci were photographed. The colonies were counted under a dissecting microscope. Five represen-tative images from each group were used to quantify foci area from three separate experiments. NIH 3T3 cells and vector-transfected NIH 3T3 cell lines were used as negative control, and the TC-1 cell line was used as the positive control.
Tumorigenesis assay and histologic analysis
NIH3T3 cell lines stably expressing HPV16 ofE6E7 or HPV16 omfE6E7 were used in a tumorigenesis assay. Cells were trypsinized, washed twice with PBS, resuspended at 5×107 cells per mL in PBS, and 0.1 mL of cell suspension was injected subcutaneously into NOD/SCID mice (8 mice in each group). All animals were monitored once a week for tumor formation. NIH 3T3 cells and vector-transfected NIH 3T3 cell lines were used as the negative controls, and the TC-1 cell line was used as the positive control.
The animals were sacrificed after xenograft tumor establishment, 5μm paraffin sections of xenograft tumors were stained with hematoxylin and eosin (H & E), three representative images from each section were obtained by light microscopic analysis after H & E staining.
-
Construction of the optimized recombinant fusion gene HPV16 ofE6E7
The synthetic gene HPV16 ofE6E7 was inserted into cloning vector pUC18. DNA sequencing assay suggested that the sequence of the synthetic ofE6E7 gene was exactly the same as designed. Further double restriction enzyme digestion on pUC18/ HPV16 ofE6E7 at Kpn Ⅰ and Xba Ⅰ produced a target band about 780bp in length (Fig. 1). This suggests that the pUC/ HPV16 ofE6E7 recombinant plasmid containing Kpn Ⅰ and Xba Ⅰ restriction sites at both ends had been constructed successfully.

Figure 1. Analysis of recombinant plasmid pUC/ofE6E7 by KpnⅠ & XhoⅠ digestion. M, DNA ladder; 1, pUC18; 2, pUC18/ HPV16 ofE6E7
Construction of expression vector
Using pUC18/HPV16 ofE6E7 as template we spliced together the optimized, mutated and fused E6E7 gene (omfE6E7) by SOE-PCR and introduced 5' BamH Ⅰ/ 3' EcoR Ⅰ restriction sites at both ends of the PCR amplified E6E7 fragment. Next, we used pcDNA3.1(+) as vector to build expression vector pcDNA3.1(+)/HPV16 ofE6E7 and pcDNA3.1(+)/ HPV16 omfE6E7. DNA sequencing verified they had been constructed correctly. Furthermore, both two recombinant plasmids were subjected to double restriction enzyme digestion, and each turned out to have the 780bp target band (Fig. 2).
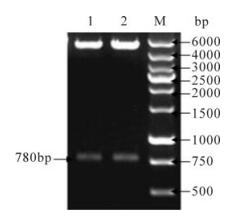
Figure 2. Analysis of the expression plasmids pcDNA 3.1(+)/ HPV16 ofE6E7 and pcDNA 3.1(+)/HPV16 omfE6E7 by BamH Ⅰ & EcoR Ⅰ digestion. 1, pcDNA 3.1(+)/HPV16 ofE6E7; 2, pcDNA 3.1(+)/HPV16 omfE6E7; M, DNA ladder
NIH 3T3 cell lines with stable transfection of ofE6E7 and omfE6E7 genes
After G418 selection, NIH 3T3 cell lines with the stable transfection of the ofE6E7 and omfE6E7 genes were identified by immunofluorescence and Western blotting. As shown in Fig. 3, both NIH 3T3-ofE6E7 and NIH 3T3-omfE6E7 cells were immunolabeled and exhibited a staining pattern in the cytoplasm. A similar immunoflourescence staining pattern was obtained with the antibody against E6 protein (data not shown). The expression of E6E7 protein in NIH 3T3-ofE6E7 and NIH 3T3-omfE6E7 cells was also examined by Western blot. As shown in Fig. 4, a distinct band was found at about 36kDa indicating the putative E6E7 protein was detected with the polyclonal antibody of anti-HPV16 E6. Densities of the Western blotting bands suggested that the expression levels of HPV16 ofE6E7 and HPV16 omfE6E7 were similar in the same amount of cells. The same result was obtained with the antibody against the E7 protein(data not shown). The results from immunofluorescence and Western blot suggested that NIH 3T3 cell lines with the stable transfection of the ofE6E7 and omfE6E7 genes were constructed successfully and both ofE6E7 and omfE6E7 genes maintained very good stability and antigenicity.
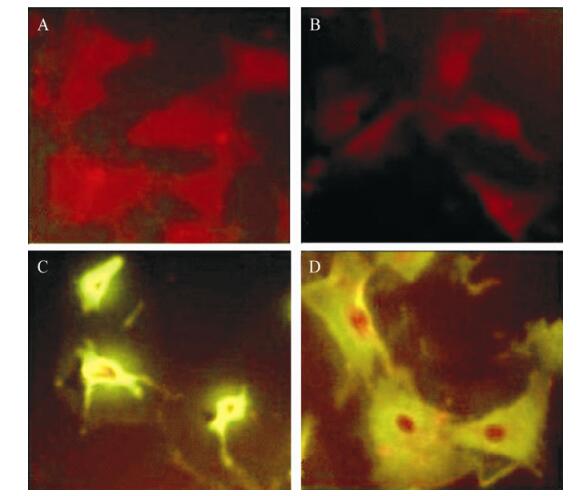
Figure 3. Immunofluorescence detection of the recombinated pcDNA expression in NIH 3T3 cells. HPV16 E7 antibody. A: NIH 3T3 Cells. B: pcDNA3.1(+). C: pcDNA3.1(+)/HPV16 ofE6E7. D: pcDNA3.1(+)/HPV16 omfE6E7

Figure 4. Western Blot detection of the recombinant pcDNA expression in NIH 3T3 cells. 1: NIH 3T3 Cells; 2: pcDNA3.1(+); 3: pcDNA3.1(+)/HPV16 ofE6E7; 4: pcDNA3.1 (+)/HPV16 omfE6E7
Identification of transforming activity
As shown in Fig. 5, after growing in soft agarose for 3 weeks, cell colonies were formed in NIH3T3-ofE6E7 cells (Fig. 5C) and TC-1 cells (Fig. 5E). However, no colony consisting of more than 50 cells was observed in NIH 3T3-omfE6E7 cells (Fig. 5D), NIH 3T3-neo cells (Fig. 5B) and untreated cells (Fig. 5A).
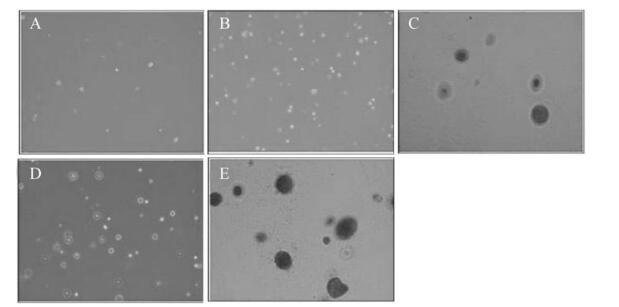
Figure 5. Colony formation of NIH 3T3 cell lines transfected with the codon optimized and recombinant HPV16 E6E7 gene in soft-agar.A: NIH 3T3. B: NIH 3T3-neo. C: NIH 3T3-ofE6E7. D: NIH 3T3-omfE6E7. E: TC-1 positive control
Tumorigenicity assays showed that the mice (n=8) injected with either NIH 3T3-ofE6E7 (Fig. 6A) or TC-1 (Fig. 6B) cells developed tumors, whereas NIH 3T3-omfE6E7, NIH 3T3-neo or NIH 3T3 cells did not cause any visible tumors (data not shown). In addition, pathological observation of the primary tumors from NIH 3T3-ofE6E7 cells revealed a malignant phenotype by hematoxylin and eosin (H & E) staining (Fig. 7). Thus, our findings reveal that engineered HPV16 omfE6E7 lost its transformation activity.
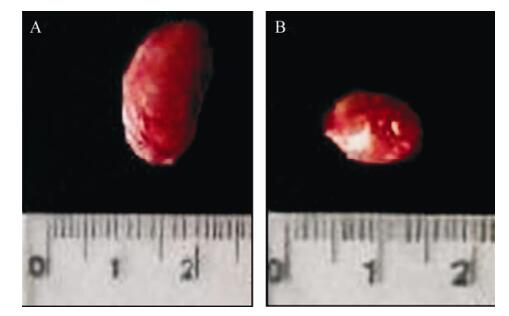
Figure 6. Tumors from SCID mice of different groups. Tumors in SCID mice in group NIH 3T3-ofE6E7 (A) and group TC-1(B)
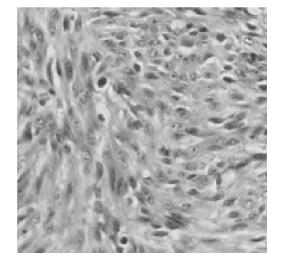
Figure 7. Micrographs of paraffin section of tumor in SCID mice in group NIH 3T3-ofE6E7
-
HPV16 E6 and E7 proteins play a major role in the development of the cervical cancer. The E6 protein can bind and degradation the p53 and destroy the normal control of the cell cycle while the E7 protein can bind the pRb protein and hence the pRb lost it ability as an anti-oncogene. The effects of the E6 and E7 proteins lead the cells to proliferate malignantly and result in cervical cancer[3, 5, 8]. Since E6 and E7 are consistently expressed in most cervical cancers and their precursor lesions but absent from normal tissues, these viral oncoproteins represent promising targets for the development of antigen-specific therapeutic vaccines for HPV-associated cervical cancer and precursor lesions.
However, analysis of the HPV genome suggests that the codons used by HPV are different from its host-human gene codons which explains the low expression level of wild-type HPV E6 and E7 genes in human cells[7]. This seriously hampers the deve-lopment of therapeutic vaccines. Secondly, although therapeutic vaccines that target either HPV16 E6 or HPV16 E7 have been previously found to induce good immune responses[4], the combination of E6 and E7 DNA vaccines may have better anti-tumor potential than either alone [5]. Thirdly, since both HPV16 E6 and E7 genes are oncogenes with potential carcinogenicity, genetic mutations at relatively oncogenic sites are necessary to eliminate their carcinogenicity to ensure their safety when used as vaccines.
Accordingly, in this work, using HPV16 E6 and E7 genes as antigen genes, we successfully optimized the wild-type HPV16 E6 and E7 genes according to the codon usage for mammalian cell expression for expecting to increase their expression level in mammalian cells, and fused the HPV16 E6 and E7 genes to include as many epitopes as possible, enabling an improved E6 or E7-specific CTL reaction. The optimized and fused HPV16 E6E7 gene has a high protein expression level and good antigenicity to antibodies against the E6 or E7 protein (Fig. 3 and 4), but it still has a transformation capability to NIH 3T3 cells (Fig. 5, 6 and 7).
Previous studies have confirmed that a single amino acid mutation such as L50G, C113R on the HPV16 E6 protein as well as C24G, E26G on the HPV16 E7 protein could effectively eliminate the transformation capability of E6 and E7 proteins[3, 6, 12]. Therefore, a mutant was generated by introduction of L50G, C113R on the HPV16 E6 protein as well as C24G, E26G on the HPV16 E7 protein to generate the HPV16 ofE6E7 gene (Patent pending (CN 101100672)). The results showed that the HPV16 omfE6E7 mutant gene maintained a high protein expression level and good antigenicity to antibodies against the E6 or E7 protein as well as the HPV16 ofE6E7 gene (Fig. 3 and 4), but lost the transformation activity to NIH 3T3 cells compared to HPV16 ofE6E7 gene (Fig. 5 and 6). It suggests that the mutant of optimized HPV16 E6E7 fusion gene (HPV16 omfE6E7) constructed in this work can be taken as a novel candidate antigen of therapeutic vaccines for HPV16-associated tumors. But future work is required to investigate the efficacy of the immunogenicity and antitumor response.

 DownLoad:
DownLoad: