











-
In the eukaryotic nucleus, the genomic DNA and histones are packaged and organized into a nucleoprotein complex called "chromatin". The fundamental packaging unit of chromatin is the nucleosome, an octamer of histones around which 147 base pairs (bp) of DNA is wrapped twice[21]. The linker histone H1 interacts with both the nucleosome core and the linker DNA, and promotes higher-order folding and compaction of chromatin (reviewed in[24]). Besides assembling DNA into chromatins to form higher-order structures in the eukaryotic nucleus, the histones also play an active role in the regulation of gene transcription through establishing a dynamic molecular interface for transcription factors and RNA polymerases to bind to DNA sequences[27].
Protamines are a group of relatively small (4.0-12.0 kDa) and structurally heterogeneous proteins. A chemical definition of the protamine can be deduced from its sequence composition of ≥40% arginine with a few or no lysine[18]. Protamines reportedly serve as functional counterparts of histones, and the remodeling from a histone-to a protamine-based chromatin will usually result in higher condensation of genomic DNA and gene transcription down-regulation (reviewed in[3]).
Besides cellular chromatin, viral chromatin also exists and plays an important role in the life cycle of many viruses (reviewed in[19]). Viruses such as simian virus 40 (SV40) and polyomavirus which use host enzymes to replicate their DNA tend to use host histones to package viral genomic DNA into virions[4]. Alternatively, for viruses such as adenoviruses using virus-encoded replication machinery, they tend to form viral chromatin via virus-encoded histone-like proteins[28].
Baculoviruses are large double-stranded DNA viruses and among them, Autographa californica multicapsid nucleopolyhedrovirus (AcMNPV) is one of the most extensively studied prototypes. The AcMNPV basic DNA-binding protein P6.9 exhibits a protamine-like amino acid composition (44% arginine and no lysine)[29]. By 10 hours post infection (hpi), P6.9 becomes associated with the viral DNA[34]. By 24 hpi, the nucleosome-like structures are completely substituted by subnucleosome-sized DNA fragments of 120 and 90 bp chromatin structure containing exclusively viral DNA[33]. As a protamine-like chromosomal protein, P6.9 was supposed to form a higher condensed chromatin and down-regulate AcMNPV gene transcription. However, Wilson et al provided evidence that the subnucleosome-sized AcMNPV chromatin is sensitive to micrococcal nuclease digestion, which is correlated to transcriptional activity[33]. This unexpected phenotype implies that P6.9's role in regulation of viral gene transcription is probably distinct from the protamines or other protamine-like proteins. However, the detailed role of P6.9 in regulation of AcMNPV gene transcription remains unknown.
Previous research demonstrates that P6.9 is essential for viral nucleocapsid assembly, but it has no influence on viral genome replication[31]. In the present study, the epigenetic role of P6.9 in regulation of AcMNPV gene transcription was characterized. We found that P6.9 as a protamine-like chromosomal protein up-regulates viral gene transcription at 12-24 hpi, which is opposite to the protamines or other protamine-like proteins that usually down-regulate gene transcription.
HTML
-
Sf9 cells were cultured at 27℃ in Grace's media containing 10% fetal bovine serum (FBS) (Gibco). AcMNPV recombinant bacmids were derived from bMON14272 (Invitrogen)[20], and propagated in Escherichia coli strain DH10B. The AcMNPV p6.9-nulled bacmid AcBacΔp6.9 was provided by Prof. Just Vlak, Wageningen University, the Netherlands[31]. And the bacmid gp64-KO which lacks of the viral envelope protein encoding gene gp64 was provided by Prof. George Rohrmann, Oregon State University, USA[30].
-
The open reading frame (ORF) of p6.9 was amplified by polymerase chain reaction (PCR) from the AcMNPV genome (strain E2) with the primers 6.9F and 6.9R (Table 1). The PCR product was then cloned into the BamH I-EcoR I sites of pFB-mCMV-eGFP[25] to generate pFB-polh-p6.9-eGFP, which was subsequently transformed to DH10B harboring bMON14272 or AcBacΔp6.9 to generate recombinant viruses Ac-p6.9eGFP and Ac-p6.9eGFPrp by the Bac-to-Bac system according to the manufacturer's protocols (Fig. 1A) (Invitrogen).

Table 1. PCR primers used in this paper
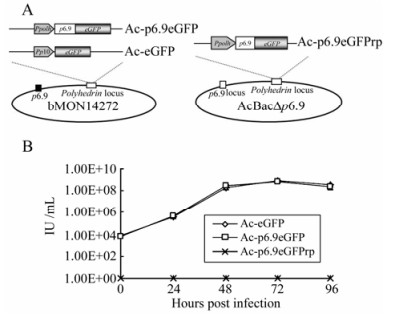
Figure 1. A: Schematic of recombinant baculovirus constructs. Three bacmid constructs were generated by using the Bac-to-Bac system. The Ac-p6.9eGFP and Ac-eGFP were produced by transposing either Ppolh-p6.9-egfp or p10-egfp expression cassettes into bMON14272, respectively. The Ac-p6.9eGFPrp was generated by transposing Ppolh-p6.9-egfp expression cassette into AcBac∆p6.9.B: Infectivity assay of the recombinant baculovirus constructs. Sf9 cells were infected with either Ac-p6.9eGFP, Ac-eGFP at 5 MOI, or supernatant from Ac-p6.9eGFPrp transfected cells at 144 hpt. The viral titers were determined by monitoring EGFP expression at the indicated time points. The data represents the averages of infections with virus from triplicate transfections, and the error bars indicate standard deviations. Note that the supernatant of Ac-p6.9eGFPrp transfected cells failed to generate any EGFP (+) cells at any indicated time points.
-
The virus infection and infectivity assay were performed as described previously[32]. Briefly, the virus stocks of Ac-p6.9eGFP and Ac-eGFP at a multiplicity of infection (MOI) of 5 or supernatant from Ac-p6.9eGFPrp transfected cells were used to infect fresh Sf9 cells. After 1 hour of infection, the viral supernatant was removed and the cells were replenished with fresh medium. Viral supernatants were collected at 0, 24, 48, 72, 96 hpi and titre was determined by endpoint dilution assay with EGFP expression as an indicator of virus infection.
-
The transcriptional patterns of viral genes were analyzed by quantitative reverse transcription-PCR (qRT-PCR). Sf9 cells plated at 5×105 per well on 6-well plates were transfected with 2 µg of AcBacΔp6.9, Ac-p6.9eGFPrp, or gp64-KO, respectively. Total RNAs were isolated by TRIzol reagent (Invitrogen) at 12 and 24 hours post transfection (hpt), and dissolved in 20 µL of RNase-free water, respectively. Total RNA (1000 ng) of each sample was digested with 1 unit of RQ1 RNase-Free DNase (Promega) for 30 min. The resultant DNA-free total RNAs in each sample (250 ng) were collected and submitted to qRT-PCR assay using a QuantiTect SYBR Green RT-PCR Kit (Qiagen) and a StepOne realtime PCR machine (Applied Biosystems). Two genes, pe38 and p10, (primer sequences are listed in Table 1) were chosen to investigate the transcriptional level of representative genes transcribed by either host RNA polymerase Ⅱ (pe38) or virus-encoded RNA polymerase (p10) and host 18s rRNA was selected as the endogenous reference. The experiment was performed in triplicate and each with three replicates.
-
Sf9 cells (5×105) were plated on slides and infected with Ac-p6.9eGFP inoculum (MOI=5). For visualization of DNA, cells at indicated time points were incubated with 100 μg/mL RNase A for 30 min at 37℃ and subsequently fixed with 4% paraformaldehyde in 0.1 mol/L PIPES, pH 6.9, 0.010 mol/L EGTA and 0.010 mol/L MgCl2 (PEM) buffer for 10 min before being permeabilized by 0.01% Triton X-100 in PBS with 2% BSA. A final incubation with 3 μg/mL propidium iodide (PI) for 2 min was then performed. For immunostaining, host RNA polymerase Ⅱ was detected with 1:500 diluted anti-ARNA-3 monoclonal antibody (Millipore) followed by addition of secondary Cy3-conjugated rabbit anti-mouse IgG (Millipore). Virus-encoded RNA polymerase was incubated with 1:500 diluted anti-LEF-8 antiserum (provided by Prof. Lorena Passarelli, Kansas State University, USA) and secondary Cy3-conjugated goat anti-rabbit IgG (Millipore). Cells were captured with a FluoView FV1000 confocal laser scanning microscope (Olympus).
-
Isolation of S1, S2, and P fractions from Sf9 nuclear chromatin was performed according to the method described previously with minor modifications[17, 26]. Nuclei of Ac-p6.9eGFP infected cells were isolated using a Nuclear Extract Kit (Active Motif) at 24 hpi, and suspended in 200 µL of nuclear buffer (20 mmol/L Tris-HCl, pH 7.5, 70 mmol/L NaCl, 20 mmol/L KCl, 5 mmol/L MgCl2, and 3 mmol/L CaCl2 supplemented with protease inhibitors). The nuclear suspension was incubated with 30 U of micrococcal nuclease (TaKaRa) at room temperature. The digestion was terminated by the addition of EDTA and EGTA 5 mmol/L of each, and the mixture was then centrifuged at 5000r/min for 3 min. This supernatant was designated the S1 fraction. The nuclear pellet was further lysed in 2 mmol/L EDTA for 15 min at 4℃, followed by centrifugation, and the supernatant and the pellet were designated as the S2 and P fractions, respectively. For Western blotting, 1:1000 diluted anti-GFP, anti-ARNA-3 or anti-LEF-8 antiserum and a 1:10, 000 diluted HRP-labeled secondary goat anti-rabbit IgG (Beyotime) were used.
-
Sf9 cells were infected with Ac-p6.9eGFP (MOI=5). At indicated time points, the cells were submitted to Chromatin immunoprecipitation (ChIP) assay using a ChIP kit (Upstate Biotechnology) and monoclonal anti-GFP antibody (Sigma) according to the manufacturer's recommendations. PCR of input virus dilutions and the bound ChIP fraction were performed simultaneously with ExTaq DNA polymerase (TakaRa). The primers used for PCR are listed in Table 1. Thirty cycles were performed and each cycle was carried out at 94 ℃ for 30 s, 55 ℃ for 30 s, and 72 ℃ for 60 s. The PCR products were separated by 2% agarose gels, visualized by staining with ethidium bromide (EB) and the bands of each PCR product were densitometrically assayed using a Gel-pro32 imager (Media Cybernetics).
-
Representative results from two to three independent experiments repeated in triplicate were shown in each figure. All experimental data values shown were calculated from triplicate samples. Data were analyzed using independent sample t-tests and were expressed as means ± standard deviation (SD), except for qRT-PCR. P-values less than 0.05 were considered significant.
Cell culture and virus
Generation of the recombinant bacmid constructs
Virus infection and infectivity assay
Quantification of viral gene transcription
Fluorescence microscopy
Chromatin fractionation
Chromatin immunoprecipitation
Statistical analysis
-
In epigenetic research, histone-enhanced green fluorescent protein (eGFP) fusion protein has proven to be a convincing and convenient tool for studying chromatin dynamics[11]. In a recent report, eGFP-tagged histone H4 was constructed to examine the host chromatin behavior in baculovirus-infected cells [23].
In the present study, the p6.9-eGFP fusion gene driven by the polyhedrin (ph) promoter was inserted into the ph locus of bMON14272 (Invitrogen), to generate the recombinant bacmid Ac-p6.9eGFP (Fig. 1A). A wild-type control bacmid, Ac-eGFP, was described previously (Fig. 1A)[32]. To evaluate whether the insertion of p6.9-eGFP at the ph locus can rescue AcBac∆p6.9 in which p6.9 was deleted[31], Ac-p6.9eGFPrp was constructed based on AcBac∆p6.9 (Fig. 1A). All of these recombinant bacmid constructs were confirmed by polymerase chain reaction (PCR) with the commercially available primer set M13± according to the Invitrogen's manual (data not shown).
To evaluate the infectivity of these bacmid constructs, a one-step virus growth curve was drawn according to the data of endpoint dilution assay. The curve demonstrated that the Ac-p6.9eGFP exhibited a similar virus growth dynamics with the wild-type control Ac-eGFP, indicating that over-expression of the P6.9-eGFP fusion protein has no influence on virus replication (Fig. 1B). Whereas the Ac-p6.9eGFPrp failed to generate infectious virions (Fig. 1B), which suggests the P6.9-eGFP fusion protein cannot fully substitute the functionality of the native P6.9.
-
To investigate whether P6.9 has an impact on viral gene transcription, qRT-PCR was performed to evaluate viral gene transcription at various infection phases. Two representative viral genes, pe38 and p10 were selected, as pe38 is an immediate early gene transcribed by host RNA polymerase Ⅱ and p10 is a very late gene transcribed by virus-encoded RNA polymerase[12, 14, 16, 22]. The gp64-KO was chosen as the wild-type control, because this gp64-nulled bacmid is deficient in virion budding, while other propagation processes like viral gene transcription should be unaffected[30].
Figure 2 showed that the transcripts of pe38 and p10 in either gp64-KO or Ac-p6.9eGFPrp transfected cells continued to increase at a similar rate from 12 to 24 hpt. However, in AcBacΔp6.9 transfected cells, the transcripts of the two representative genes remained at the same level at 12 and 24 hpt. These two distinct transcriptional phenotypes demonstrated that P6.9 can unanimously up-regulate viral gene transcription in the late infection phase (i.e. 12-24 hpt), regardless of the source of the RNA polymerase used. Also P6.9 appeared to be non-essential for the basal level of viral gene transcription, as the viral gene transcripts can still be detected at 12 hpt in the cells transfected with AcBacΔp6.9 that lacks p6.9.

Figure 2. Quantification of AcMNPV gene transcription in Sf9 cells transfected with gp64-KO, Ac-p6.9eGFPrp, or AcBac∆p6.9, respectively. The transcription levels of pe38 and p10 were normalized to host 18s rRNA transcripts. The transcription level of each gene at 12 hpt is configured as 1. Bars represent means and standard errors of the means for three independent experiments, and the asterisks (*) indicates p < 0.05.
Additionally, the similar transcription accumulation rate in gp64-KO and Ac-p6.9eGFPrp transfected cells indicated that the P6.9-eGFP fusion protein can functionally substitute the native P6.9 to restore viral gene transcription, and is thus valid to characterize P6.9 in terms of viral gene transcription regulation.
-
Since our data demonstrated that P6.9 is involved in viral gene transcription regulation and P6.9 is a protamine-like chromosomal protein that may epigenetically influence viral gene transcription[34], it is necessary to investigate whether the P6.9-DNA association is temporally and spatially synchronized with P6.9's impact on viral gene transcription.
After Spodoptera frugiperda (Sf9) cells were infected with Ac-p6.9eGFP, P6.9-eGFP was located in the cytoplasm and formed discrete dots by 8 hpi (Fig. 3). At 16 hpi, P6.9-eGFP was diffusely distributed throughout the infected cell, primarily in the nucleus region and partially co-localized with the propidium iodide (PI)-stained DNA (Fig. 3). By 24 hpi, as the viral DNA accumulated, P6.9-eGFP became completely co-localized with DNA in the virogenic stroma region (Fig. 3). The time course of P6.9-DNA co-localization matched well with the P6.9's impact on viral gene transcription at 12-24 htp, and suggested that P6.9's association with viral genomic DNA is crucial for its functionality in regulation of viral gene transcription.
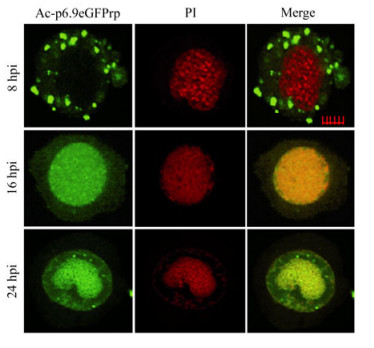
Figure 3. Dynamic subcellular distribution of P6.9. Sf9 cells were infected with Ac-p6.9eGFP (MOI=5). The subcellular distribution of P6.9eGFP was captured at 8, 16, and 24 hpi. The nuclear DNA is PI-stained.
-
We have demonstrated P6.9 can up-regulate viral gene transcription at 12-24 hpi, and the onset of P6.9-DNA co-localization is temporally and spatially matched with this regulation process. In order to determine whether P6.9-DNA association contributes to the viral gene transcription up-regulation, a chromatin fractionation assay based on limited micrococcal nuclease digestion was performed[17, 26].
Chromatin fractionation showed P6.9 was present in all three fractions, and was particularly enriched in the pellet fraction P and supernatant fraction S1 at 24 hpi (Fig. 4A). This phenotype suggested that the P6.9-DNA complex was mostly concentrated in the nuclear matrix binding fraction P that consists of nuclease-resistant chromatin including actively transcribed gene sequences, and the fraction S1 that harbors open genetically active chromatin as demonstrated with a papilloma virus[17, 26].
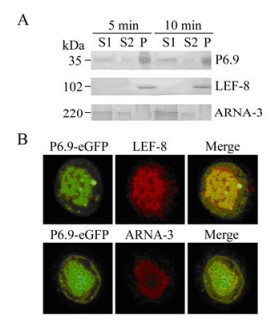
Figure 4. P6.9 is associated with transcriptionally active chromatin. A: Chromatin fractionation assay. Ac-p6.9eGFP was used to infect Sf9 cells. At 24 hpi, cells were subjected to chromatin fractionation assay, and the nuclei were fractionated into S1, S2, and P fractions. The abundance of P6.9, virus-encoded RNA polymerase (represented by LEF-8), and host RNA polymerase Ⅱ (represented by ARNA-3) in each chromatin fraction was detected by Western-blotting with different exposure time (5 and 10 min). B: Sf9 cells were infected with Ac-p6.9eGFP (MOI=5). At 24 hpi, cells were incubated with anti-ARNA-3 or anti-LEF-8 antiserum and detected with a Cy3 conjugated secondary antibody.
Also, the AcMNPV-encoded RNA polymerase subunit LEF-8 was exclusively enriched in fraction P[8], whereas host RNA polymerase Ⅱ was primarily presented in fraction S1, as manifested by anti-ARNA-3 antibody (Fig. 4A). Note that the anti-ARNA-3 antibody can specifically react with amino acids 794-822 of the largest RNA polymerase Ⅱ subunit in humans as well as in insects according to Millipore's product description. However, this amino acid sequence is lacking AcMNPV-encoded RNA polymerase subunits LEF-4, LEF-8, LEF-9, and P47 based on the sequence analysis (Data not shown), which indicates that ARNA-3 cannot serologically cross-react with AcMNPV-encoded RNA polymerase.
Accordingly, the immunofluorescence microscopy of Ac-p6.9eGFP infected cells at 24 hpi revealed that P6.9 co-localized with LEF-8 throughout the virogenic stroma region (Fig. 4B), indicating this region is abundant in chromatin fraction "P". P6.9 also co-localized with the host RNA polymerase Ⅱ at the periphery of the virogenic stroma region (Fig. 4B), suggesting that this region is where the chromatin fraction "S1" is enriched and undergoing active transcription.
Therefore, our data suggested that in the late infection phase, the transcriptionally active chromatin fraction where P6.9 is enriched not only acts as the transcription templates for virus-encoded RNA polymerase, but also for host RNA polymerase Ⅱ.
-
To further characterize the P6.9 association with viral DNA, which is essential for P6.9's role in up-regulating viral gene transcription, a detailed study of the P6.9-viral DNA binding status in the course of virus infection was carried out.
To pull down the p6.9-DNA complex from Ac-p6.9 eGFP infected cells, chromatin immunoprecipitation (ChIP) with anti-GFP was performed. The precipitated DNA sequences were then submitted to semi-quantitative PCR using the primer sets designed to amplify the selected promoters belonging to viral immediate early, delayed early, and late genes, as well as the hr sequences (primer sequences were listed in Tab. 1)[6, 7]. Typical examples of the PCR products amplified from the precipitated chromatin are shown in Fig. 5A. In general, the results demonstrated that the levels of P6.9 binding to various viral DNA sequences started to increase from 8-12 hpi. And by 24 hpi, the P6.9's enrichments at various viral DNA sequences accumulated to a similar level without statistical difference as manifested by the densitometry assay (Fig. 5B), suggesting that P6.9's binding to viral DNA is sequence-independent and in accordance with the qRT-PCR results that either pe38 or p10 transcripts were unanimously up-regulated upon P6.9's presence at 12-24 hpt. Note that at 48 hpi, all the immunoprecipitated DNA fragments decreased in comparison with 24 hpi. This is probably because the majority of viral genomic DNA will be packaged into nucleocapsids at 48 hpi, while P6.9-eGFP is absent from the nucleocapsids.

Figure 5. Characterization of the association between P6.9 and viral DNA. A: Sf9 cells were infected with Ac-p6.9eGFP (MOI=5). At indicated time points, the cells were submitted to ChIP using anti-GFP antibody to pull down P6.9-eGFP and its associated DNA. Various PCR primers that amplify viral genes or hr sequences (listed in Tab. 1) were used to examine the abundance of viral DNA associated with P6.9 in the immunoprecipitates. The semi-quantification results as represented by EB-stained DNA bands are shown in "CH" column. The input viral DNAs were simultaneously assayed by PCR (displayed in "IN" column). The white arrow indicates the correct hr2 PCR band. B: Densitometry assay of relative P6.9 enrichment at various viral genomic fragments. The PCR bands shown in A were converted to densitometric data by Gel-Pro32. The relative enrichment of P6.9 is presented as log2 of the PCR product in the ChIP sample divided by that in the input sample (log2[CH/IN]). The data reflected the mean value of triplicate measurements ± SD.
Additionally, the binding of P6.9 to hr sequences appeared to be slightly prior to the other viral genomic fragment tested. This phenotype suggested the hr sequences as the viral transcriptional enhancers may probably act as the earliest binding target for P6.9 to trigger the up-regulation of viral gene transcription[6, 7].
Generation and infectivity assay of the recombinant bacmid constructs
P6.9 up-regulates viral gene transcription in the late infection phase
The co-localization of P6.9 and DNA is temporally and spatially synchronized with P6.9's impact on viral gene transcription
P6.9 and RNA polymerase are enriched in the transcriptionally active chromatin fraction
Characterization of P6.9-viral DNA association
-
Chromatin remodeling is an important pathway to regulate gene transcription epigenetically. The role of protamine as a chromosomal protein in suppression of cellular gene transcription has been extensively documented (reviewed in[3]). However, in this research we examined P6.9, an AcMNPV protamine-like chromosomal protein that exhibits an opposite effect in regulation of viral gene transcription in contrast to other identified protamines or protamine-like proteins.
Besides P6.9's function in up-regulating gene transcription, one of the major differences between P6.9 and cellular protamines appears to be their genomic distribution pattern. Not only is the heterogeneous distribution of histones over the genome a common feature among eukaryotic cells (reviewed in[35]), but also cellular protamines bind to genomic DNA in a sequence-dependent manner, as manifested by their selective enrichment over β-globin and Alu sequences in human sperm nucleus[36]. In this research, the AcMNPV genome-wide distribution of P6.9 was investigated. In contrast to cellular protamines, ChIP suggested that P6.9 binds to various genomic regions ranging from viral immediate early to late promoters in a sequence-independent manner. This was consistent with a previous report which found that both active and relatively inactive genes of AcMNPV produce similar proportions of subnucleosomal-sized DNA fragments upon micrococcal nuclease digestion[33].
Virus-encoded protamine-like protein has been previously described. The adenovirus major core protein Ⅶ is recruited to the nucleus at the early infection phase, and functions as a transcriptional suppressor through condensation of viral genomic DNA[10]. In comparison with P6.9 structurally, the adenovirus protein Ⅶ possesses four basic domains, including one protamine-like and three histone-like domains segregated by alpha helices[28], whereas P6.9 contains only a single protamine domain. Whether this structural discrepancy contributes to their functional difference in regulating viral gene transcription needs further exploration.
According to our data, host RNA polymerase Ⅱ is enriched and co-localized with P6.9 at the periphery of the virogenic stroma region (Fig. 4B), where the chromatin fraction "S1" undergoes active transcription in the late infection phase (Fig. 4A). It is reasonable to hypothesize that P6.9 probably contributes to viral gene transcription up-regulation in the late infection phase by binding to and presenting viral DNA to host RNA polymerase Ⅱ at the periphery of the virogenic stroma region, where host RNA polymerase Ⅱ transcribes viral immediate early genes, which subsequently trans-activate downstream genes in a cascade manner[13].
Actually, incidences of viral chromosomal protein interacting with host transcriptional machinery is not unusual. As mentioned previously, adenovirus protein Ⅶ, as a viral chromosomal protein, indirectly interacts with host transcriptional factors through binding to E1A[10], which is reportedly in association with several host transcriptional factors, including ATF2, TATA box binding protein (TBP), TBP-associated factors, and the mammalian Srb-Mediator complex (reviewed in[5]).
In addition, these findings also prompt us to re-examine the definition of the baculovirus expression phase. The classical expression phase is defined based on the time point before or after the viral DNA replication, the RNA polymerase encoded by the host cell or the virus, and the different types of promoters. However, from an epigenetic perspective, the formation of de novo synthesized virus-specific subnucleosome may also be considered as a criterion to define the virus infection phase. Analysis of AcMNPV transcription kinetics indicates that all the 155 viral genes are at high transcriptional levels in the late infection phase[9]. Dai et al revealed that several early, late and very late Helicoverpa armiger NPV (HearNPV) genes start to transcribe within their own phases, and unanimously reach their highest level at 72 hpi[2]. In contrast, the HearNPV genome replication reaches its highest level at 14-20 hpi, and begins to decline until it essentially ceases at 60 hpi. These desynchronized phenotypes suggest that the highest transcription level at 72 hpi is not entirely due to the increase of virus genome copy number. We think one of the possible scenarios is that irrespective of the temporal phase, late in infection the transcriptional templates of all genes are P6.9-based subnucleosomal chromatin that exhibits a higher transcriptional activity than nucleosomal chromatin.
In summary, our data provides a novel instance that AcMNPV-encoded protamine-like protein P6.9 up-regulates viral gene transcription, and host RNA polymerase Ⅱ may probably contribute to this up-regulation process in the late infection phase. In addition, P6.9 appears to bind to various viral genomic fragments in a sequence-independent manner, which suggests that P6.9 acts more like a transcriptionally active chromosomal protein rather than a transcription factor to up-regulate gene transcription epigenetically.

 DownLoad:
DownLoad: