









-
Cyanobacteria ("blue-green algae") are ancient oxygenic photosynthetic organisms that are widespread in the environment, and even constitute the dominant primary producer in extreme permafrost environments of aquatic systems (Jungblut A D, et al., 2005; Sherman L A, et al., 1970). However, mass developments of cyanobacteria in lakes and brackish waters, called "algal blooms" or "water blooms", have been a serious concern due to their frequent association with toxins, which pose a health hazard to humans, domestic animals and wildlife (Carmichael W W, 2001; Codd G A, et al., 2005; Dittmann E, et al., 2006; Lorraine C B, 2002). Until recently, the mortality of phytoplankton due to virus infection has been shown to play an important role in the fading of algal blooms. Moreover, viruses are considered to greatly impact microbial population dynamics and ecosystems (Fuhrman J A, 1999; Sabehi G, et al., 2012; Suttle C A, 2005, 2007).
A series of reports have indicated that viruses are quite abundant, diverse, and ubiquitous in the aquatic environment (Bergh O, et al., 1989; Bratbak G, et al., 1990; Proctor L M, et al., 1990; Suttle C A, 2007). As agents that infect cyanobacteria, cyanophages have attracted significant interest. Since the first cyanophage LPP-1 was isolated in 1963 (Safferman R S, et al., 1963), a number of new cyanophages have been isolated and characterized from both fresh and sea waters (Chen F, et al., 2002; Liu X Y, et al., 2007; Liu X Y, et al., 2008; Mann N H, et al., 2005; Pope W H, et al., 2007; Safferman R S, et al., 1963.). Based on morphology, all tailed cyanophages are classified into three families of double-stranded DNA viruses (Safferman R S, 1983): Myoviridae (contractile tails), Siphoviridae (long non-contractile tails) and Podoviridae (short tails).
In the past, most investigations focused on the fundamental biological characteristics of the cyanophages until the last decade when, in 2002, the total genome sequence of cyanophage P60 (Chen F, et al., 2002) was reported. Since then, the genomes of several cyanophages (Gao E B, et al., 2012; Liu X Y, et al., 2008; Mann N H, et al., 2005; Sabehi G, et al., 2012; Sullivan M B, et al., 2005; Weigele P R, et al., 2007) have been sequenced and analyzed, which has contributed to investigations on the biological characteristics and virus/host co-evolutionary relationships(Sullivan MB, et al. 2009). The number of totally sequenced genomes was also sufficient to form the basis of a taxonomic approach. Yet, in spite of the ubiquitous distribution of cyanophages, the number of totally sequenced genomes is actually quite small. The genomes revealed several relatively conserved structural genes and also carry many genes of unidentified function (Raytcheva D A, et al., 2011).
Cyanophage PP, used in this study, was the first cyanophage isolated in China (Zhao Y J, et al., 2002) and can specifically infect the filamentous cyanobacterium Plectonama boryanum and Phormidium foveolarum. PP has been the subject of many investigations since 2002. According to Liao (Liao M J, et al., 2010), cyanophage PP is a short-tailed, icosahedral-shaped, double-stranded DNA virus, which can be frequently detected in high abundance in many eutrophic lakes in China.
In order to better understand the genetic properties of cyanophage PP and to uncover the interactions between the cyanophage and its hosts, we sequenced its complete genome and investigated its fundamental biological characteristics.
HTML
-
The cyanobacterium used in this study was Plectonema boryanum, obtained from the Freshwater Algae Culture Collection of the Institute of Hydrobiology, Chinese Academy of Sciences. Clonal axenic cultures were routinely grown in BG11 medium at 28 ℃ under fluorescent lights, which were maintained on a 12 h on / 12 h off cycle. For long-term storage, a concentrate of P. boryanum cells was maintained on an oligotrophic BG11 agar plate (0.7% agar) and then kept under dim light.
-
PP was kindly provided by Zhao et al. (2002). In our study, P. boryanum was cultured to 107 - 108 cells/mL and infected with cyanophage PP at a multiplicity of infection (MOI) of 5 - 10. On days 3 - 4 post infection, the culture turned from green to yellow, indicating lysis of the host cells. Progeny cyanophages were stabilized by the addition of MgSO4 (final concentration 20 mmol/L) to the lysate followed by treatment with 2% (vol/vol) chloroform for 5 - 10 min. Cell debris were removed by centrifugation at 10000×g for 10 min at 4 ℃. The supernatant was layered onto a 30% sucrose cushion and centrifuged at 50, 000 rpm (Ty 70 rotor; Beckman Coulter, CA, USA) for 1 h. The pellets were resuspended in 0.1×TE buffer (pH 7.4), loaded onto a 15 - 60% continuous sucrose density gradient, and centrifuged at 35, 000 rpm (SW 41 rotor, Beckman Coulter) for 1.5 h. Each band was collected separately with sterile needles and syringes, mixed with 0.1×TE buffer, and centrifuged at 40, 000 rpm (SW 55 rotor, Beckman) for 1 h to wash away the sucrose. The pellets were individually resuspended in fresh 0.1×TE buffer and the virus stocks were immediately used or stored at -80 ℃ until needed.
-
A freshly purified cyanophage suspension (10 μL) was layered onto a carbon-coated copper grid. Excess sample was blotted off 5 min later and the particles were negatively stained with 2% uranyl acetate for 1 min. The excess stain was removed with a filter paper. Finally, the samples were examined with a Tecnai G2 20 TWIN transmission electron microscope (FEI Company, OR, USA) at 200 kV.
-
Double-agar overlay plaque assay was used to determine the titer of cyanophage PP, as described by Kropinski et al. (2009), with some modifications. Briefly, 10 mL of 2% BG11 agar medium (underlay) was dispensed onto 90 mm Petri plates and 0.7 % BG11 agar medium (overlay) was distributed into 15 mL sterile tubes (3 mL per tube) and, when cool, bagged and stored at 4 ℃. Serial dilutions of cyanophage were prepared in BG11. Tubes containing 3 mL overlay medium were placed in boiling water to melt the agar and kept in a 48 - 50 ℃ water bath until use. Samples of 100 μL of each dilution were mixed with 900 μL of P. boryanum cells in the exponential phase (108 - 109 cells/mL), kept at room temperature for 5 min, transferred to tubes with 3 mL warmed overlay medium, mixed and poured onto the underlay plate. Plaque-forming units (PFUs) were counted after 40 - 48 h incubation under the same conditions used to grow the host cells.
-
P. boryanum was cultured in BG11 medium and grown to a density of 1 to 2×107 cells/mL, centrifuged at 7000×g for 15 min, and the pellet was resuspended in fresh medium. The next day, PP stocks were added to give a MOI of 1, and incubated with shaking at 28 ℃ in the dark for 30 min to allow the phage to adsorb. The mixture was centrifuged and the infected cells were washed twice to remove unadsorbed phage and incubated with fresh warm BG11 medium. During a 96 h infection period, supernatants containing extracellular progeny phage, were collected every 24 h. The titers of all samples were determined as described above. Three independent phage titrations were carried out and the average for each time point was plotted.
-
Genomic DNA was extracted for sequencing as described previously (Wilson W H, et al., 1993). Briefly, the phage was propagated as above and, after the addition of 20 mmol/L MgSO4 and 1 mol/L NaCl, the lysates were placed at 4 ℃ for 1 h. The samples were treated with chloroform and centrifuged to remove cell debris. Polyethylene glycol 8000 (10%, wt/vol) was added to the supernatant to precipitate phage particles, which were then dissolved in 2×lysis buffer (20 mmol/L Tris/HCl, 20 mmol/L EDTA, 0.5% SDS; pH 7.6), treated with 3 μL proteinase K (20 mg/mL) and incubated at 58 ℃ for 2 h. An equal volume of phenol-chloroform-isoamyl alcohol (25:24:1) was added to the phage suspension and the suspension was gently vortexed and centrifuged, then the aqueous layer was removed. This procedure was performed twice. Subsequently, CH3COONa (final concentration 0.3 mol/L, pH 7.0) and isopropanol (equal volume) were used to precipitate DNA, which was then washed twice by 70% ethanol and resuspended with sterile water. The PP genome was sequenced by the shotgun-sequencing method. Basically, genomic DNA was digested by several restriction endonucleases, and the small fragments were cloned into pUC19 and sequenced. ContigExpress (NY, USA) was employed to assemble these sequences into contigs, and gaps were filled using a primer-walking technique.
Potential open reading frame (ORF) prediction was performed by using GeneMark, Glimmer and ORF Finder software. Translated ORFs were used to search for homologs by BLASTp against the non-redundant database and Swiss-Prot protein database in NCBI.
-
The restriction enzymes Xba Ⅰ, Hind Ⅲ, EcoR Ⅰ, Cla Ⅰ, Kpn Ⅰ, Nde Ⅰ and Spe Ⅰ were used to digest PP DNA, as recommended by the supplier (Takara). The reactions, containing 1.5 - 2.0 μg of DNA and enzyme (20 U), were incubated at 37 ℃ overnight to allow complete digestion. They were stopped by adding 10×loading buffer. Gel electrophoresis was carried out in 0.6% agarose gel at 35 V for 3 - 4 h to ensure that most of the bands were well separated.
Host alga growth conditions
Cyanophage propagation and purification
Electron microscopy
Cyanophage titration
One-step growth curve
DNA extraction and genome analysis
Restriction endonuclease digestion
-
As reported earlier (Zhao Y J, et al., 2002), PP is a short-tailed, icosahedral-shaped, double-stranded DNA virus, 52 nm in size, which infects the filamentous cells of Plectonema boryanum and Phormidium foveolarum. However, previous studies were not clear about the tail part of the phage. In an attempt to gain more detail about the structure and morphology of the phage, we purified the cyanophage by sucrose density gradient ultracentrifugation. Four major bands, 1#(15%~30%), 2#(40%~50%), 3#(50%~55%) and 4#(55%) were observed. Negative staining revealed that most of the bands contained cell debris except band 3, which had pure cyanophage particles. Band 2 contained filamentous material and a few empty particles. However, even in band 3, most of the particles were empty shells without nucleic acid; this may have been due to the instability of cyanophage PP.
Electron micrographs of negatively stained PP (Fig. 1A) revealed icosahedral virions (diameter approx. 52 nm), each with a stubby short tail. Tail fibers and other typical phage structures were not visible in this study. Morphologically, cyanophage PP belongs to the family Podoviridae. Our finding is corroborated by other investigators (Zhao Y J, et al., 2002).
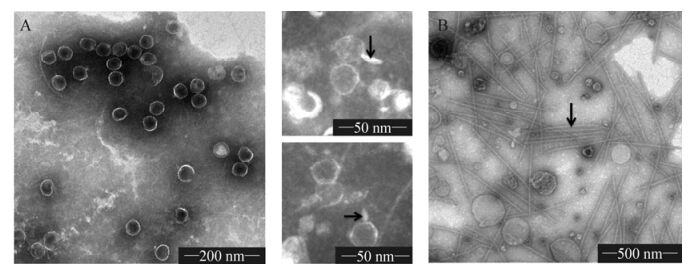
Figure 1. Electron micrographs of cyanophage PP purified by sucrose density gradient ultracentrifugation. A, electron micrograph of purified PP virions (arrows indicate the short and stubby tail structure); B, filamentous artifacts (arrow) observed in the purification of PP.
The unknown artifacts in Fig. 1B were numerous and relatively uniform in size - about 800 nm in length, and 15 nm in diameter. In comparison with the flexible flagellae and cilia, they were rigid and resembled certain plant viruses and filamentous phages, such as TMV and M13 phage.
-
The double-agar overlay plaque assay was applied to determine the concentration of infectious phage particles (Fig. 2). Fig. 2B shows that many sharply defined round and clear plaques were formed on a lawn of P. boryanum and without halo, which is a hallmark of temperate phages and also a distinct difference between lysogenic and lytic phages (Paul J H, et al., 2010), indicating that cyanophage PP is a virulent virus. Furthermore, Fig. 2A shows that the serial dilutions are well reflected by the number of plaques on the plates and that the plaques became larger with increased dilutions (Fig. 2B). Apparently, as cells grow, more phages are propagated and diffuse into the new cells (Zhao Y J, et al., 2002).
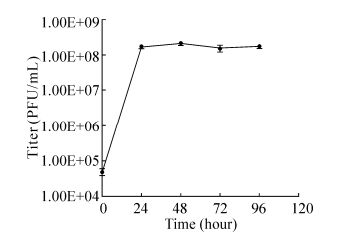
Figure 2. One-step growth curve of PP infection of the cyanobacterium P.boryanum. Each data point represents the average results of three individual experiments; bars denote the standard deviation.
During frequent subculture and long-term preservation of PP, some mutants could be generated. Here, we demonstrated the use of double-agar plaque assay to purify the cyanophage. Following three rounds of purification and propagation, the titer of PP increased from the initial 104 to 107 - 108 PFU/mL.
The one-step growth curve of PP in Figure 2 showed that the phage titer reached the maximum (108 PFU/mL) 24 h post infection, and remained stationary for the following 72 h, revealing that the host cells were almost completely lysed by cyanophage 24 h post infection. However, owing to the long interval between the two time points, the latent period was not detected.
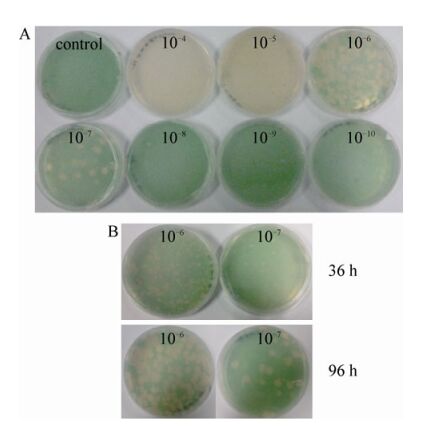
Figure 3. Double-agar overlay plaque assay to measure the titer of PP. A, the appearance of plaques formed by cyanophage PP after a series of dilutions; B, the appearance of plaques formed by cyanophage PP after 36 h and 96 h incubation.
-
The genomic fragment sequences of PP were assembled into a circular contig. However, regarding the terminal redundancy that occurs in many phages or cyanophages such as T4, Ma-LMM01 and S-PM2, genomic DNA was digested by certain enzymes to identify whether the PP genome was circular or linear. Based on theoretical restriction enzyme analysis of the whole genome sequence of cyanophage PP, Cla Ⅰ, Kpn Ⅰ, Nde Ⅰ and Spe Ⅰ were selected for their proper segments. Theoretically, cyanophage PP has nine Cla Ⅰ restriction sites, one Kpn Ⅰ restriction site, nine Nde Ⅰ restriction sites and two Spe Ⅰ restriction sites. According to the data presented in Fig. 4, both the number and size of the bands reflect a linear genome. Since the number of bands equalled one more than the number of restriction sites, the genome of cyanophage PP was shown to be a linear molecule, not a circular one.

Figure 4. Digestion of PP genomic DNA with Cla Ⅰ, Kpn Ⅰ, Nde Ⅰ and Spe Ⅰ. The first lane is λ Marker (λ DNA digested by EcoR Ⅰ, BamH Ⅰ and Hind Ⅲ), and the last one is DL5000 Marker.
-
The genome of cyanophage PP is a 42, 480 bp, linear, double-stranded DNA molecule with 222 bp terminal repeats and a G+C content of 46.41%. The linear genome and the terminal repeats were confirmed by enzyme digestion and PCR fragment sequencing (data not shown). The genome sequence was submitted to GenBank and the access number is KF598865. It has high similarity with the known Pf-WMP3 sequence. The genome has 41 ORFs packed closely to occupy 93.1% of the genome, 17 of which have homologs in other phages or microbes. Table 1 summarizes the results and lists the putative functions and properties of the ORFs. The nucleotide positions, orientations, sizes and probable functions of all 41 ORFs are displayed in Fig. 5, which shows that the genome can be divided into two different parts, designated part A and part B. The first 23 ORFs in part A are oriented to the right, while the next 18 ORFs in part B are oriented to the left. Among the 17 ORFs with assigned functions, 13 were transcribed leftward, encoding structural proteins such as the major capsid protein (ORF33), tail protein (ORF25, ORF26), tail tube (ORF29, ORF31) and tail fiber (ORF24). Apart from these structural proteins, part B also contains scaffold protein, portal protein and terminase, also found in other phages of the T7 supergroup. However, homologs to genes on part A are rarely found in the current databases, and two (DNA primase/helicase and DNA polymerase) of the four relatively conserved genes which encode proteins necessary for DNA replication and modification.
ORF Position Length a Predicted protein Related phage(s) or microbes E valueb 1 419-685 88 2 774-1334 186 3 1331-1567 78 4 1545-1748 67 5 1949-3202 417 6 3292-3837 181 7 3956-4624 222 8 4624-5133 169 9 5078-5371 97 10 5377-6636 419 11 6641-8815 724 DNA primase/helicase Pf-WMP3, P60, Syn5, P-SSP7, K1F, Pseudomonas phage phi15, vibriophage VP4, phiYeO3-12, T3, T7 0.0 12 8876-9076 66 13 9089-9295 68 14 9299-11167 622 DNA polymerase Pf-WMP3, Pf-WMP4, Marinithermus hydrothermalis DSM 14884 0.0 15 11149-11304 51 16 11497-11742 81 17 11826-12788 320 18 13017-13193 58 19 13272-13514 80 20 13522-14025 167 ERF family protein(phage-associatedrecombination protein) Nostoc flagelliforme str. Sunitezuoqi 1e-26 21 14038-14247 69 22 14210-14548 112 23 14627-15487 286 gp17(exchanged genefrom other phages) Roseobacter phage SIO1 2e-18 24 16753-15551 400 Tail fiber Pf-WMP3, Enterobacter cloacae subsp. Cloacae ATCC 13047, P2 0.0 25 20201-16740 1153 Tail fiber Pf-WMP3 0.0 26 25437-20299 1712 Tail protein Pf-WMP3, Pf-WMP4 0.0 27 28469-25485 994 Endopeptidase(M23/M37) Pf-WMP3, Pf-WMP4, Cyanothece sp. PCC 7424, Lyngbya majuscula 3L 0.0 28 29661-28471 396 Internal protein Pf-WMP3 0.0 29 32748-29665 1027 Tail tube B Pf-WMP3 0.0 30 33103-32738 121 Recombinationendonuclease Ⅶ Streptomyces sp. e14, vibriophage ICP2_2006_A, vibriophage VP4, vibrio phage phiYeO3-12, S-PM2, Mycobacterium phage L5 5e-15 31 33773-33108 221 Tail tube A Pf-WMP3 5e-164 32 34166-34050 38 33 35361-34216 381 Capsid Pf-WMP3, Pf-WMP4, Deep-sea thermophilic phage D6E, uncultured phage MedDCM-OCT-S04-C24, Cyanophage NATL2A-133, NATL1A-7, P-SSP7 0.0 34 36067-35384 227 Scaffolding Pf-WMP3 6e-165 35 38027-36072 651 Portal Pf-WMP3, Pf-WMP4, Roseobacter phage SIO1, T3, T7 0.0 36 38467-38042 141 DNA endonuclease Methanopyrus kandleri AV19 0.22 37 40201-38504 565 Terminase Pf-WMP3, Pf-WMP4, Synechococcus phage S-CBS2, Cyanophage 9515-10a, NATL1A-7, Vibrio phage VP93, T7 0.0 38 40807-40307 166 39 41298-40813 161 40 41594-41319 91 41 41850-41599 83 a Amino acid. b Each putative ORF was used to search for homologs by BLASTp against the non-redundant database and Swiss-Prot protein database in the NCBI. The phage or microbe with the most closely related sequence is listed first and that. value is reported. Table 1. ORFs of PP genome and their presumed functions
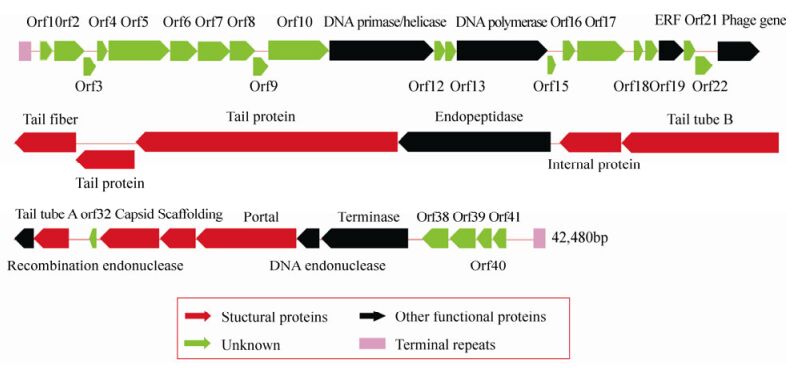
Figure 5. Genome organization of cyanophage PP. Arrows indicate the size, position and orientation of 41 ORFs, and the different colors represent different functions of the ORFs.
Purification and electron microscopy of PP
Characterization of PP infection
Identification of genome structure
Genome organization
-
We investigated the fundamental characteristics and genome sequence and organization of the first cyanophage isolated in China. The genome of cyanophage PP is a 42, 480 bp, linear, double-stranded DNA molecule with 222 bp terminal repeats. Morphologically, PP is a 52 nm icosahedral virion, with a stubby short tail. Previous reports have substantially missed the tail structure (Liao M J, et al., 2010; Zhao Y J, et al., 2002), but our EM studies clearly show the short and stubby tail structure.
In the process of purification of cyanophage PP with the method of sucrose density gradient ultracentrifugation, numerous filamentous artifacts were observed. Even after three times purification by a double-agar plaque assay process, they were still not removed. The presence of such artifacts may indicate a virus that is closely related to cyanophage PP; however, such a relationship between PP and the putative virus remains to be confirmed. However, the persistent existence of these artifacts is not likely to be a coincidence because filamentous objects have been discovered in Synechococcus spp. infected with a cyanophage (Prof. Shi Zhengli, personal communication). The presence of such possible plant viruses may be indicative of an evolutionary relationship between cyanobacteria and plants.
Intriguingly, the PP genome could not be digested by commonly used enzymes, such as EcoR Ⅰ, BamH Ⅰ, Not Ⅰ, Nhe Ⅰ and Nco Ⅰ, which indicated that the restriction sites were modified by methylation or another process so as to protect the phage genome from being digested by the restriction endonuclease in its host cells. As different cyanobacteria may have varied restriction endonucleases, this may partly account for the host specificity of cyanophages.
The genome was shown to contain 41 potential ORFs which can be divided into two distinct parts, differing both in orientation and function. Genes in the conserved part B appear to function mainly in virion assembly, whereas the diverse genes in part A are primarily needed for genome replication. However, certain special genes that are generally found in cyanophages could not be detected in the PP genome, for example: psbD (Sullivan M B, et al., 2005), psbA (Chenard C, et al., 2008) genes related to the photosynthesis of cyanophages, the NblA gene (Yoshida T, et al., 2008b), which is essential for the degradation of the phycobilisomes, g20 (Wilhelm S W, et al., 2006), g91 (Yoshida M, et al., 2008a) and MazG (Bryan M J, et al., 2008) genes. Unique genes in the PP genome did not have homologs in the current database and may have functions that contribute to the genetic diversity of microbial communities.
Cyanophages play an important role in the disappearance of water bloom, and in our study, we investigated a virulent cyanophage that can lyse P. boryanum and P. foveolarum. Although P. boryanum and P. foveolarum are not the main cyanobacteria that induce water blooms, our work has provided a platform for investigating other cyanophages. In the future we shall focus on isolating and investigating further cyanophages that can infect cyanobacteria involved in water blooms, such as Microcystis and Anabaena. We may also undertake the synthesis and modification of cyanophages to expand their host range and induce a specific function.
-
We are grateful to Professor Zhengli Shi and Genfu Xiao for their helpful advice. Professor Lirong Song is acknowledged for providing cyanobacteria and Associate Professor Kai Cheng for providing cyanophage PP.
-
Zhou Yiran wrote the manuscript and finished the PP purification and genome sequencing; Lin Juan conducted the PP infectivity investigation; and Li Na helped the genome sequence analysis and annotation. Hu Zhihong advised on the project design and execution and on the writing of the manuscript. Deng Fei was the corresponding author who designed the project and undertook the writing and editing of the manuscript.

 DownLoad:
DownLoad: