









-
Viruses are the most abundant biological entities on the earth (Suttle C A, 2005). In marine ecology, viral lysis contributes up to 70% of cyanobacterial mortality and 90 - 100% of bacterial mortality in freshwater systems, suggesting that viruses play key roles in the control of microbial communities (Fischer U R, et al., 2002; Proctor L M, et al., 1990; Weinbauer M G, et al., 1998).
Fresh water is an important ecosystem which presents an activity interface between humans and a variety of host organisms. Freshwater lakes and ponds are drinkwater sources and also a habitat for water plants and the larval stage of many insects, and may serve as a medium for virus intra-and interspecies transmission. In China, nearly all major inland freshwater lakes represent different levels of human-induced eutrophication over past decades (Qin B, 2002; Song L, et al., 2007). Eutrophication of freshwater lakes has led to cyanobacterial harmful blooming, which has caused a drinking water crisis and human health threat. The occurrence of heavy cyanobacterial blooming in many large lakes in warm seasons has increased in frequency and intensity in recent years. Meanwhile, cyanobacterial genera such as Microcystis and Anabaena can produce toxins, like hepatotoxins, that can cause liver failure in wild animals, livestock, and aquatic life as well as human illnesses (Carmichael W W, 2001; Peng L, et al., 2010). East Lake is a typical eutrophic freshwater lake located in Wuhan city, China. Algal blooming in this lake has occurred frequently over the last decades. A recent study by transmission electron microscope (TEM) revealed a high abundance of planktonic viruses in East Lake, which could reach up to 109 viral particles per milliliter of water (Liu Y M, et al., 2006). However, little is known genetically about these viruses. One cyanophage (PaV-LD) that infects harmful filamentous cyanobacterium Planktothrix agardhii has been isolated from East Lake, but until now, no cyanophages infecting major blooming cyanobacteria in fresh water, like Microcystis and Synechococcus, have been reported (Gao E B, et al., 2012). In marine water, cyanophages infecting Prochlorococcus and Synechococcus are extremely abundant. Considering the number of viruses and bacteria is almost ten-fold higher in fresh water than in marine environments, we can therefore expect a much higher abundance of cyanophages in fresh water (Lu J, et al., 2001; Maranger R, et al., 1995; Sullivan M B, et al., 2003).
Most viruses in the environment cannot be cultured in the laboratory, which limits our knowledge of virus diversity. High-throughput sequencing (HTS) techniques, independent of virus culture, have revealed viral diversities in several ecosystems, including marine water, fresh water, animal feces, and human feces (Angly F E, et al., 2006; Cantalupo P G, et al., 2011; Djikeng A, et al., 2009; Lopez-Bueno A, et al., 2009; Phan T G, et al., 2011; Zhang T, et al., 2006). In this study, we collected four samples representing four seasons, and we applied random amplification followed by Solexa sequencing to each sample. Our results revealed high virus diversity and abundance changes in different seasons.
HTML
-
Four water samples were collected in Aug-2009, Dec-2009, Mar-2010, and June-2010, respectively. One hundred liters of surface water ( < 0.5 m in depth) were collected from four locations within East lake (DH1, 30°32′54.78″N, 114°21′14.43″E; DH2, 30°32′45.43″N, 114°22′1.35″E; DH3, 30°32′22.98″N, 114°23′28.43″E, DH4, 30°32′48.03″N, 114°25′23.16″E), which were labeled on a map (Supplementary Fig. 1). Water physical and chemical characteristics covering the four specified months were collected from the Hubei Environmental Protection Bureau, which were summarized in Supplementary Table 1. Virus-like particles were concentrated, following the protocols described by Thurber RV et al. (2009). Briefly, 100 L water were piped through a 2 μm pore size filter, then through a 0.45 μm pore size filter. The filtrates were concentrated to 400 mL by the tangential-flow filtration method using a 50 kD filter, and finally concentrated to 5 mL by ultracentrifugation through a 20% sucrose cushion at 80, 000 g for 90 min using a Ty90 rotor (Beckman Coulter, Brea, CA). The pelleted viral particles were dissolved in 400 μL phosphate-buffered saline (PBS). PBS-dissolved viral particles were negative stained and observed under TEM.
-
The concentrated viral particles were treated with DNase (Fermentas, part of Thermo Fisher Scientific, Waltham, MA), benzonase (Novagen, part of Merck KGaA, Darmstadt, Germany), and RNase (Fermentas) to digest unprotected nucleic acid at 37 ℃ for 60 min. Viral nucleic acids were extracted using the QIAamp viral RNA kit (Qiagen, Limburg, Netherlands) following the manufacturer's instructions. Viral nucleic acids was eluted in 60 μL AVE buffer (Qiagen, Limburg, Netherlands), and stored at - 80 ℃. Viral nucleic acid libraries containing both DNA and RNA viral sequences were constructed by sequence-independent RT-PCR amplification, as previously described by Ge X et al. (2012). A 10 pmol random primer Brs (GCCGGA GCTCTGCAGAATTCNNNNNNNN) containing a 20-base fixed sequence at the 5' end followed by a randomized octamer at the 3' end (8N) was used in a reverse transcription reaction with murine leukemia virus reverse transcriptase (Promega, Madison, WI). A single round of DNA synthesis was then performed using Klenow fragment polymerase (Takara, Kyoto, Japan), followed by PCR amplification of nucleic acids using primers consisting of only the 20-base fixed portion of the random primer Bs (GCC GGA GCT CTG CAG AAT TC) with KOD-Plus DNA polymerase (Toyobo, Osaka, Japan).
-
The random PCR products were loaded in 2% agarose gel, and fragments of appropriate size (500 - 1, 500 bp) were purified from gel with a Gel Extraction Kit (Omega Bio-Tek, Norcross, GA). For each sample, 2 μg of random PCR products were barcoded, then pooled together for the Solexa (part of Illumina, San Diego, CA) sequencing by the BGI (Shenzhen, China).
-
Solexa reads were initially generated and assigned to four data sets based on the different barcodes. Clean reads of 75 bp were obtained after removing the adaptors, barcodes, fixed primers (20 bp), and eight additional nucleotides (encoded by the 8N part of the random primers). Trimmed sequences within each group were assembled into contigs using Soapdenovo software, with a criterion of > 95% identity over at least 15 bp. Assembled contig sequences were compared with GenBank data using Blastn and Blastx. Using a Blastx search, sequences with e-values of ≤ 10-5 were classified as likely originating from a eukaryotic virus, bacterium, phage, eukaryote, or unknown based on the taxonomic origin of the sequence with the best e-value.
-
Specific primers (Supplementary Table 2) targeting the conserved sequences of capsid assembly protein gene (g20) (Sullivan M B, et al., 2008; Zhong Y, et al., 2002), photosystem Ⅱ core reaction center proteins D1 gene (psbA), photosystem Ⅱ core reaction center proteins D2 gene (psbD) (Chenard C, et al., 2008; Sullivan M B, et al., 2006), and DNA primase-helicase gene (Wang K, et al., 2004) were used for cyanophage detection. The viral total DNA/RNA from four samples was used as a template. The PCR products were gel purified using a Gel Extraction Kit (Omega Bio-Tek, Norcross, GA) according to the manufacturer's instructions and cloned into the pGEM-T Easy Vector (Promega, Madison, WI) before sequencing. For each PCR fragment cloned, three independent clones were sequenced to obtain the consensus sequence.
-
Routine sequence management and analysis were carried out using DNASTAR (Madison, WI) software or Geneious software (Biomatters Ltd, Auckland, New Zealand). Sequence alignment and edit were performed using ClustalW, BioEdit, or GeneDoc. Phylogenetic trees were constructed using the maximum likelihood algorithm with bootstrap values determined by 1, 000 replicates in the MEGA5 software package. High-throughput data sets were deposited in MG-RAST under ID numbers 4535211.3, 4535210.3, 4535208.3, and 4535209.3. Detected new g20, psbA, psbD, and DNApol gene sequences of cyanophage were deposited in GenBank under the following accession numbers: KF709158 - KF709192.
Water sample collection and viral-like particle concentration
Viral nucleic acid purification and sequence-independent PCR amplification
Library construction and Solexa sequencing
Sequencing data assembly and processing
Detection of cyanophages in water samples
Sequence analysis and nucleotide sequence accession numbers
-
Viral particles concentrated and purified from water samples were observed under TEM and showed diverse morphology. Some of them are similar to siphoviruses and myoviruses (Fig. 1).

Figure 1. Morphological diversity of viruses in Lake Donghu observed under transmission electron microscopy. S, siphoviruses; M, myoviruses.
-
High-quality Solexa clean reads were assembled to contig sequences; in total, 234, 669, 71, 837, 13, 820, and 34, 236 contigs (> 90 bp) were obtained from samples of Aug-2009, Dec-2009, Mar-2010, and Jun-2010, respectively. Contig sequences were classified using a Blastx homology search against the GenBank non-redundant (GenBank_nr) database. Blastx top matches with e-value ≤ 10-5 were used to carry out the species annotation. The Blastx analysis showed that most of the contig sequences (88.9 - 91.6%) had no significant homology to any known sequences with assigned species. This phenomenon is similar with other published viral metagenomic data and reveals our limited knowledge of biodiversity in the natural environment. The remaining contigs (8.4 - 11.1%) could be assigned to bacteria, virus, eukaryote, and Archaea species (Fig. 2; Supplementary Table 3). From this analysis, we found that the total contig number of each sample was quite different, possibly owing to the cDNA library quality; alternatively, the higher biodiversity (hosts and viruses) in Aug-2009 may also have contributed to more assembly contigs. But, we found that the percentages of contigs classified as unknown species in every sample were similar (~90%).
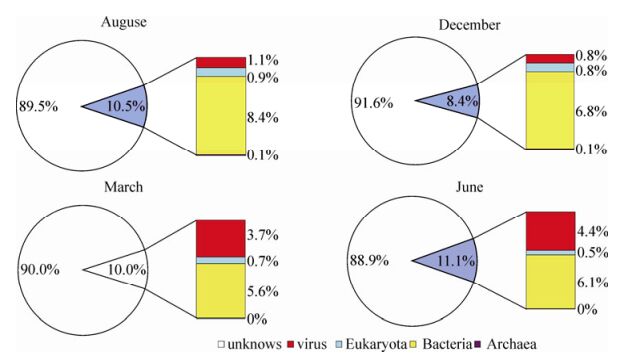
Figure 2. Compositionof contigs homologous to viruses, bacteria, Eukaryota, and Archaea in four samples collected from East lake.
-
Based on the best Blastx matching, contigs homologous to known virus sequences were classified into 23 viral families, including 21 DNA viral families and 2 RNA viral families (Fig. 3, Supplementary Table 4). Meanwhile, many contigs were found to be homologous to unclassified viruses (not assigned to any known viral families) in each sample (Supplementary Table 4). In terms of the classification of the contigs, there may be three reasons for the inequality between the DNA and RNA viral families. First, RNA viruses may occur significantly less than DNA viruses in East Lake water. Second, the RNA viruses were more sensitive to pressure and destroyed by the filtration step. Third, the rPCR step may have amplified the RNA viruses less efficiently. The majority of contigs were homologous to dsDNA (double stranded DNA) viruses, including myoviruses, podoviruses, siphoviruses, and phycodnaviruses, which infect bacteria, prokaryotic algae (Abedon S T, 2009; Ackermann H W, 1998; Van Duin J T N, 2006), or eukaryotic algae (Wilson. W H, et al., 2005). The second dominant contigs were homologous to ssDNA (single stranded DNA), including microviruses that infect bacteria and circoviruses that infect invertebrates and vertebrates (Delwart E, et al., 2012; Marvin D A, 1990; Roux S, et al., 2012). Few sequences were found to be homologous to parvoviruses (densovirus and bocavirus) (Hueffer K, et al., 2003), plant-infecting nanoviruses and geminiviruses (Grigoras I, et al., 2012; Muhire B, et al., 2013), fish-and amphibian-infecting alloherpesviruses (Waltzek T B, et al., 2009), iridoviruses (Williams T, et al., 2005), baculoviruses, nimaviruses, polydnaviruses (Escobedo-Bonilla C M, et al., 2008; Kelly B J, et al., 2007; Turnbull M, et al., 2002), ascoviruses (Federici B A, et al., 2009), herpesviruses, adenoviruses, poxviruses (Benko M, et al., 2003; Davison A J, 2002; Hughes A L, et al., 2010), mimiviruses (Claverie J M, et al., 2009), and lipothrixviruses (Prangishvili D, et al., 2004). Virus species changes were observed with the dominant sequences homologous to dsDNA viruses in August and December, and ssDNA in March and June.
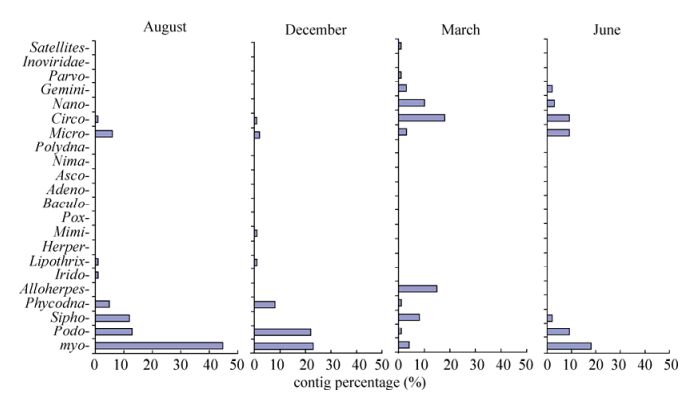
Figure 3. Relative abundance of contigs homologous to viral families.
-
Cyanophage sequences homologous to members of Myoviridae, Podoviridae, and Siphoviridae were detected in samples of the four seasons, with the most abundant in Aug-2009, and the least abundant in Mar-2010, reflecting diversity and abundance changes, possibly associated with physical factors and host community (Table 1).Myoviruses are contractile-tailed phages which were found to be widespread in global marine environments in viral metagenomic surveys (Angly F E, et al., 2006; Williamson S J, et al., 2008). In this study, we found that the majority of contigs were homologous to myoviruses, including P-SSM2 and P-SSM4, which have been found to infect oceanic primary producers Prochlorococcus (Sullivan M B, et al., 2003), S-RIM2, S-RIM17, and Syn9 in another major oceanic primary producer Synechococcus (Marston M F, et al., 2012). However, most cyanophagerelated sequences detected in our study showed low amino acid identities (around 50%) with these marine cyanophages, indicating that these freshwater myoviruses represent novel cyanophages. A high proportion of sequences have a high similarity (82 - 100% nucleic acid identities) to a myovirus Ma-LMM01, which was isolated from Lake Mikata in Japan in 2006 and can specifically infect a toxic strain of the algal-bloom-forming cyanobacterium Microcystis aeruginosa (NIES298 strain) (Yoshida T, et al., 2006). Sequence alignment showed that 337 contigs were mapped and with a coverage of 37% in comparison with the Ma-LMM01 full-length genomic sequence (162 kb) (Fig. 4). This result suggests that there is one (or more) microcystis phage similar to Ma-LMM01 in East Lake, leading to the possibility of isolating this virus, which can lyse toxic microcystis. Several contigs homologous to members of Podoviridae were detected, including P-SSP7, which infects oceanic Prochlorococcus, and S-CBP2, S-CBP3, P60, and Syn5, which infect Synechococcus (Labrie S J, et al., 2013; Raytcheva D A, et al., 2011). All these contigs showed less than 80% amino acid identities with known cyanophages, indicating that there are novel podoviruses in East Lake. In addition, a few sequences homologous to siphovirus PSS2, which has been detected in oceanic Prochlorococcus, were also detected in our Solexa sequencing data.
Viral family and species Solexa PCR-cloning Aug Dec Mar June Aug Dec Mar June Myoviridae 789 50 13 57 16 37 0 1 Prochlorococcus phage P-SSM2 100 8 2 11 0 0 0 0 Prochlorococcus phage P-SSM4 77 12 2 18 14 6 0 0 Synechococcus phage S-RIM2 0 2 0 0 0 0 0 0 Synechococcus phage S-RIM17 1 0 0 0 0 0 0 0 Synechococcus phage S-RIM24 1 0 0 0 0 0 0 0 Synechococcus phage S-RIM50 1 0 0 0 0 17 0 0 Synechococcus phage S-BnM1 4 1 0 0 1 3 0 1 Synechococcus phage Syn9 71 4 2 11 0 0 0 0 Synechococcus phage S-RSM4 86 15 5 6 0 0 0 0 Synechococcus phage S-PM2 73 3 2 11 0 10 0 0 Synechococcus phage S-WHM1 5 0 0 0 0 0 0 0 Synechococcus cyanophage Syn1 0 0 0 0 0 1 0 0 Microcystis phage Ma-LMM01 370 5 0 0 1 0 0 0 Podoviridae 19 25 0 1 0 0 0 0 Prochlorococcus phage P-SSP7 9 14 0 0 0 0 0 0 Synechococcus phage S-CBP2 0 1 0 0 0 0 0 0 Synechococcus phage S-CBP3 1 0 0 0 0 0 0 0 Synechococcus phage P60 2 1 0 0 0 0 0 0 Synechococcus phage Syn5 7 9 0 1 0 0 0 0 Siphoviridae 4 10 0 0 0 0 0 0 Marine cyanobacterial siphovirus PSS2 4 10 0 0 0 0 0 0 Unclassified cyanophages 11 11 0 0 0 1 0 0 Phycodnaviridae 134 48 6 4 Acanthocystis turfacea chlorella virus 52 23 8 2 Paramecium bursaria chlorella virus 53 10 1 1 Emiliania huxleyi virus 10 7 0 0 Ostreococcus tauri virus 12 7 1 0 Acanthocystis turfacea chlorella virus 52 23 8 2 Pyramimonas orientalis virus 0 0 0 1 Unclassified phycodnavirus 7 1 0 0 Total 1009 167 31 64 16 38 0 1 Table 1. Numbers of contigs or sequences homologous to cyanophage and phycodnavirus obtained by Solexa and PCR-cloning sequencing techniques

Figure 4. Mapping of 337 contigs from an Aug-2009 sample compared with that of the Microcystis phage Ma-LMM01. Genome of Ma-LMM01 indicated by blue lines; contigs indicated by black lines.
-
With generic primers targeting conserved genes, including capsid assembly protein (g20), photosystem Ⅱ core reaction center proteins D1 (psbA), photosystem Ⅱ core reaction center proteins D2 (psbD), and DNA primase-helicase gene (DNApol), a number of different sequences were detected in samples by PCR (Table 1). Similar to the Solexa data, the majority of sequences were detected in samples collected in Aug-2009 (16 different cyanophage sequences) and Dec-2009 (38 different cyanophage sequences), with only one cyanophage sequence detected in June-2010, and no cyanophage sequence in Mar-2010.
Degenerated g20 primers were known to amplify cyanophages of the family Myoviridae, but not Podoviridae, Siphoviridae, or other bacteriophages (Zhong Y, et al., 2002). Using these primers, seven different g20 sequence fragments (592 bp) (named as DH2009Dec-1 to 7) were detected in the Dec-2009 sample, but not in the other samples. These seven sequences could be classified into four groups according to their amino acid (aa) identities: group1 (DH2009Dec-1), group 2 (DH2009Dec-2, 3, and 4 with 96 - 99% aa identities), group 3 (DH2009Dec-5), and group 4 (DH2009Dec-6 and 7 with 98% aa identities). The aa identities between groups were less than 62% (data not shown). Alignment showed that low aa identities ( < 84%) were found with the known g20 sequences detected in fresh and marine water. On the phylogenetic tree, all the cyanophage sequences detected in this study were grouped into known cultured or uncultured freshwater cyanophages (Fig. 5). In detail, DH2009Dec-1 was closer to cyanophages (CUL02M and CUL02H) found in Cultus Lake, Canada (Short C M, et al., 2005); group 2 to a cyanophage (LAC95A) in Lake Constance, Germany (Wang K, et al., 2004); group 3 to a cyanophage (PFW-CM17) in floodwater in Japan; and group 4 to PFW-NoF2 and PFW-CF9 in floodwater in Japan (Short C M, et al., 2005). Most cyanophage sequences detected in this study are similar to those detected in environmental freshwater samples, and only a few sequences are similar to isolated cyanophages that infect marine Synechococcus or Prochlorococcus. Thus we deduced that these East Lake cyanophages may infect freshwater Synechococcus or Prochlorococcus (Sullivan M B, et al., 2010; Zhong Y, et al., 2002).
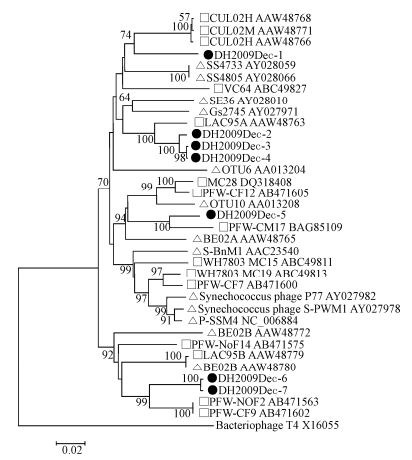
Figure 5. Phylogenetic analysis of cyanophage g20 protein sequences. Partial g20 gene sequences were amplified from a sample collected in Dec-2009. The translated g20 sequences (181 amino acids) were aligned with those of known freshwater and marine cyanophages and used for phylogenetic tree construction. Black dot, sequences obtained in this study; square, cyanophages from fresh water; triangle, cyanophages from marine water. Bacteriophage T4 g20 sequence was used as an outgroup. Cyanophage sequence information used in the phylogenetic analysis is listed in Supplementary Table 6.
Primers targeting the conserved motifs of two core photosystem reaction center genes, psbA and psbD, in myoviruses and podoviruses were used to screen the four samples (Sullivan M B, et al., 2006), and gene fragments (776 bp and 590 bp) were only amplified from the Dec-2009 sample. Twenty-eight fragments were confirmed by sequencing, 19 of which were homologous to psbA sequences and 9 to psbD sequences. The 19 psbA fragments, showed 82 - 94% aa identities among themselves and showed < 88% aa identities with known cyanophages. Among the partial psbA sequences, 17 (DH2009Dec-8 to 24) were clustered to Synechococcus myovirus strain S-RIM50, S-SM1, S-RIM2, Syn-syn19, S-SM2, S-RSM2, S-WHM1, and Syn-SPGM99-20 (Angly F E, et al., 2006; Chenard C, et al., 2008; Marston M F, et al., 2012; Sullivan M B, et al., 2008; Sullivan M B, et al., 2010), one (DH2009Dec-25) to Synechococcus myovirus strain Syn-Syn1, S-SSM2, and S-ShM2 (Marston M F, et al., 2012; Sullivan M B, et al., 2008; Sullivan M B, et al., 2010), and one (DH2009Dec-26) to Synechococcus myovirus S-PM2 (Sullivan M B, et al., 2010) (Fig. 6). The nine psbD sequences, showed 96 - 100% aa identities among themselves and showed 65 - 97% identities with known cyanophages. On the phylogenetic tree, nine psbD sequences formed a distinct branch from those found in Synechococcus (S-RSM2, S-RIM49, Syn-syn9, Syn-syn10, and Syn-syn28) (Angly F E, et al., 2006; Chenard C, et al., 2008; Sullivan M B, et al., 2010) and Prochlorococcus (P-SSM1) (Sullivan M B, et al., 2006) (Fig. 7). These results indicated that genetically diverse cyanophages infecting Synechococcus existed in East Lake, particularly in December.

Figure 6. Phylogenetic analysis of cyanophage psbA genes. Nineteen partial psbA sequences (650bp) were amplified from a sample collected in Dec-2009. The sequences were aligned with other known freshwater and marine cyanophages and used for phylogenetic tree construction. Black dot, sequences obtained in this study; square, Synechococcus myoviruses; triangle, Prochlorococcus myoviruses; diamond, Synechococcus podoviruses; circle, Prochlorococcus podoviruses; inverted triangle, cyanobacteria. Cyanophage sequence information used in the phylogenetic analysis is listed in Supplementary Table 6.
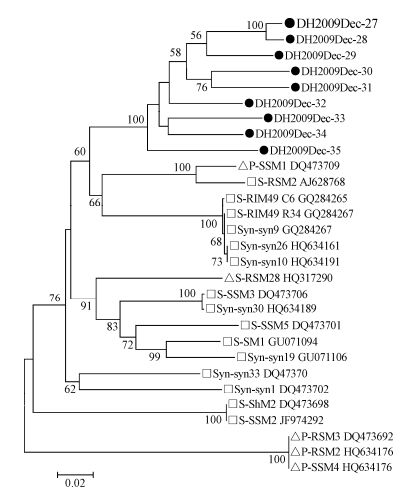
Figure 7. Phylogenetic analysis of cyanophage psbD genes. Nine partial psbD sequences (590 bp) of cyanophage were amplified from a sample collected in Dec-2009. The sequences were aligned with known freshwater and marine cyanophages and used for phylogenetic tree construction.Black dot, sequences obtained in this study; square, Synechococcus myoviruses; triangle, Prochlorococcus myoviruses. Cyanophage sequence information used in the phylogenetic analysis is listed in Supplementary Table 6.
We screened the four samples using one pair of degenerated primers targeting the conserved domain (555 bp) of the primase-helicase gene (DNApol) of the cyanophages' genomes (Sullivan M B, et al., 2006), and 14 different DNApol sequences (DH2009Aug-1 to 14) were amplified only from the Aug-2009 sample. The predicted amino acid sequences translated from these partial 14 DNApol genes, and one sequence homologous to DNApol genes obtained from Solexa data (DH2009Augcontig233468) had high sequence identities - from 88 to 98% - among each other, but low identities ( < 58%) with other known cyanophage sequences. Phylogenetic analysis showed that the 15 partial DNApol genes formed a distinct branch and had long genetic distance with known cyanophages infecting Synechococcus and Prochlorococcus (Fig. 8). These results indicated that there was a distinct lineage of cyanophages in East Lake with great genetic diversity.

Figure 8. Phylogenetic analysis of cyanophage.DNA primase-helicase protein sequences. Fifteen partial.DNA primase-helicase genes were amplified from a sample collected in Aug-2009. The translated protein sequences (181 amino acids) were aligned with those of known cyanophages and used for phylogenetic tree construction. Black dot, sequences obtained in this study; square, Synechococcus cyanophage; triangle, Prochlorococcus cyanophage. Bacteriophage T4 phage DNApol sequence was used as an outgroup. Cyanophage sequence information used in the phylogenetic analysis is listed in Supplementary Table 6.
-
Members of the Phycodnaviridae family, which infect eukaryotic algae in both marine and fresh water, have large dsDNA genomes of 150 - 400 kb. In this study, by HTS, we detected sequences homologous to five different phycodnaviruses, predominantly in the Aug-2009 and Dec-2009 samples (Table 1). These phycodnaviruses include Acanthocystis turfacea chlorella virus and Paramecium bursaria chlorella virus, which infect freshwater chlorella (Fitzgerald L A, et al., 2007; Yanai-Balser G M, et al., 2010), and Emiliania huxleyi virus, Ostreococcus tauri virus, and Pyramimonas orientalis virus, which infect different marine algae. All detected sequences showed low aa identities with these known viruses (24 - 86%), suggesting that the detected sequences in East Lake are novel phycodnaviruses.
-
In our Solexa data, 388 (7.5% of the total viral sequences) circular ssDNA sequences were found in our Solexa data. These sequences were homologous to members of families Circoviridae, Geminiviridae, and Nanoviridae, which have been reported in aquatic environments previously. These sequences exclusively matched to a conserved domain (pfam2407) of replication-related genes and were distantly related to known ssDNA viruses (data not shown). The abundance of ssDNA virus sequences allowed us to identify the replication protein and stem loop of circular ssDNA genomic elements, which are highly conserved among nanoviruses and circoviruses (Supplementary Table 5). According to their distinct phylogenetic position, we suggest that these viruses may belong to novel circular ssDNA viral families. However, because these viruses originated from environmental water samples, the host range remains unclear.
Viral particles in East Lake
Solexa sequencing, reads assembly, and BLAST analysis
Overview of viral sequences in East Lake
Cyanophages in East Lake
Genetic diversity of cyanophages detected by PCR
Phycodnaviruses in East Lake
Diverse circular ssDNA genomes in East Lake
-
East Lake is a eutrophic freshwater lake and algal blooms emerge frequently. Earlier studies of virus quantity statistics in East Lake indicated high virus abundance and seasonal variation changes in the lake (Liu Y M, et al., 2005; Liu Y M, et al., 2006). In this study, we demonstrated for the first time, from genomic information, a high genetic diversity of algal viruses in this water system. Interestingly, the majority of viral sequences in our Solexa data are homologous to DNA viruses, with the dominant dsDNA viruses (myoviruses, podoviruses and phycondanviruses) in Aug-2009, Dec-2009, and June-2010, and the dominant ssDNA viruses (circoviruses, geminiviruses, and nanvoviruses) in Mar-2010 and June-2010. The number of contigs homologous to bacteriophage microviruses remains high in Aug-2009 and June-2010. The observed transition from dsDNA virus to ssDNA virus and dynamic changes among different viral families over time possibly reflect a seasonal shift in host organisms. Similar viral shift phenomena were also found in an Antarctic freshwater lake (Lopez-Bueno A, et al., 2009). Meanwhile, the limit and bias of the available annotated sequences in the GenBank database may cause bias of organism annotation in our data. The fact that most of sequences in East Lake are homologous to bacteriophages, cyanophages, phycodnaviruses, and plant viruses further demonstrated that the DNA viruses in this water system play important roles in cycling carbon and nutrients through control of host cell communities, as well as in shaping microbe evolution by supplying the host with new genetic material as gene transfer agents (Sullivan M B, et al., 2006; Suttle C A, et al., 1994).
Among the contigs homologous to viruses, a high proportion was homologous to marine cyanophages that infect Microcystis, Synechococcus, and Prochloron; all of these algae were algal bloom species in East Lake, particularly Microcystis aeruginosa (Zhou J Z C, Wang L L., 2009). In the Aug-2009 sample, almost half of the myovirus-related sequences (370 of 789 contigs) showed high homologous (82 - 100% nucleic acid) identities to a myovirus Ma-LMM01, which was isolated from Lake Mikata in Japan in 2006 and infected a toxic strain of Microcystis aeruginosa (NIES298 strain) (Yoshida T, et al., 2006), which led to the possibility of isolating a strain similar to Ma-LMM01 in East Lake. The high proportion of Ma-LMM01-related sequences in the Aug-2009 sample is consistent with the high level of blooming caused by Microcystis aeruginosa in the summer.
Using degenerated primers targeting the conserved domain of g20, psbA, psbD, and DNApol gene, genetically diverse cyanophage sequences were amplified from the Dec-2009 sample, then from the Aug-2009 and June-2010 samples, but not from the Mar-2010 sample. These results were consistent with the Solexa data for the DNApol gene, but not for other three genes in which most cyanophage sequences were detected in the Dec-2009 sample. The disparity between the Solexa data analysis and PCR may be caused by the specific selection of primer, which preferentially amplifies certain clusters of phage sequences which are popular in the winter (Sullivan M B, et al., 2006). Generally, based on the phylogenetic analyses of amplified partial sequences of g20, psbA, psbD, and DNApol genes, we found that cyanophages in East Lake had relatively long evolution distance with known cyanophages. Consistently with Solexa data analysis, most sequences detected by PCR should represent novel fresh water cyanophages.
In addition to cyanophages, we have also detected a number of sequences that have low similarity to eukaryotic algae-infecting phycodnaviruses, including Acanthocystis turfacea chlorella virus, Paramecium bursaria chlorella virus, Emiliania huxleyi virus, Ostreococcus tauri virus, and Pyramimonas orientalis virus. Acanthocystis turfacea chlorella virus and Paramecium bursaria chlorella virus infect Chlorella-like green algae that are not the main algal blooming species in fresh water; the number of these viruses and their hosts should be positively correlated (Yamada T, et al., 2006). The other three related viruses were marine algae-infecting viruses; the native freshwater hosts of these similar viruses still need further exploration (Larsen J B, et al., 2008).
In summary, by HTS and PCR, we detected a large number of genetically diverse cyanophage and phycodnavirus sequences in a eutrophic freshwater lake, East Lake, and revealed the seasonal diversity and abundance changes of planktonic viruses in this lake. Our results provide useful viral genetic information which expands our knowledge of the freshwater virome and will be helpful for future virus isolation and control of algal blooming.
-
We acknowledge financial support from the Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-YW-Z-0954, KSCX2-EW-Z-3).
-
Zhengli Shi designed and coordinated the study. Xingyi Ge, Yongquan Wu, Lijun Wu, Xinglou Yang, and Yuji Zhang collected the samples. Xingyi Ge and Yongquan Wu conducted the majority of experiments. Xingyi Ge, Yongquan Wu, Jun Wang, and Meiniang Wang analyzed the data. Xingyi Ge and Zhengli Shi wrote the manuscript.
-
The supplementary materials are available on the website of Virologica Sinica: http://www.virosin.org.

 DownLoad:
DownLoad: