











HTML
-
Tobacco mosaic virus (TMV) is a positive-sense single-stranded RNA virus belonging to the Tobamovirus family (He M, et al., 2012). TMV infects at least 100 different species, particularly tobacco, causing huge production losses. The virus is transportable over long distances and infects crops through contaminated water used for irrigation (Tosic M, et al., 1984). In 1994, TMV spread due to contaminated water resulted in significant production losses in tobacco in the Fujian Province of China (Zheng Y T, et al., 2000). A s ensitive and specific method to detect virus contamination and monitor irrigation water is therefore necessary for crop protection.
To date, numerous methods have been applied for TMV detection. A direct enzyme-linked immunosorbent a ssay (ELISA) was initially developed for detection of TMV (Bar-Joseph M & Saloman R, 1980). However, this method was not sufficiently sensitive to allow detection of TMV at low concentrations. Recently, an immunocapture reverse transcriptase-polymerase chain reaction (IC-RT-PCR) protocol was developed and used widely to detect several fruit tree viruses (Wetzel T, et al., 1992; Rowhani A, et al., 1995). IC-RT-PCR was subsequently applied for the detection of tomato mosaic virus (ToMV) and TMV (Jacobi V, et al., 1998). Compared with ELISA, IC-RT-PCR has a number of advantages, such as higher sensitivity, specificity and effectiveness. However, the method is not sufficiently sensitive to facilitate detection of TMV within large volumes of water. In 2010, the one-step reverse transcription loop-mediated isothermal amplification (RT-L AMP) assay was developed to detect TMV (Liu Y, et al., 2010). Although RT-LAMP led to significantly enhanced sensitivity, the technique could not be used to quantify TMV. Real-time PCR, developed in 1996 based on the conventional PCR method, has been extensively used for virus detection because of its rapidity, specificity and sensitivity (Bertolini E, et al., 2008). Compared with the other available methods, real-time PCR offers significant advantages in detecting disease agents, validating its utility as a reliable diagnostic tool.
In general, TMV is present at extremely low concentrations in water and difficult to detect directly using real-time PCR. Therefore, concentration of the virus from water samples is necessary for detection (Boben J, et al., 2007). Several techniques for concentrating aquatic viruses from large volumes have been developed and successfully applied in the past. The nucleic acid method is considered a sensitive and specific approach for virus detection (Abbaszadegan M, et al., 1993) but disruption of ribonucleic acid polymerase activity is a major limitation. Phenol-chloroform extraction, sodium dodecyl sulfate-proteinase K digestion, guanidinium thiocyanate extraction and trichloroacetic acid precipitation have additionally been developed to detect waterborne viruses (Jiang X, et al., 1992; Yang F, et al., 1993; Shieh Y S C, et al., 1995). However, these methods lead to exposure of viral nucleic acids, particularly single-stranded RNA, causing degradation before analysis. The macromolecular compound, PEG, is commonly used as a precipitating agent (Chun P W, et al., 1967). PEG was initially used to concentrate and purify Bacteriophage T2 and Adenovirus (Philipson L, et al., 1960). Since then, PEG has been applied for the detection of Hepatitis A virus, Poliovirus type 1, and Influenza A virus in water (Jaykus L A, et al., 1996; Schwab K J, et al., 1996; Deboosere N, et al., 2011). The PEG-based method has been proved to be rapid, inexpensive and applicable under various conditions to concentrate virus from water samples (Colombet J, et al., 2007). These findings collectively support the utility of PEG as a suitable concentration agent for TMV in water.
According to previous studies, PEGs with a molecular weight (MW) below 6000 are less effective in precipitating macromolecules, while those with molecular weight equaling and above 6000 appear more effective and behave identically (Colombet J, et al., 2007). In the current study, PEG6000 was selected to concentrate TMV from water samples, and a TMV-specific real-time PCR method optimized to facilitate virus detection in the concentrate.
-
N. tabacum plants were inoculated with TMV under greenhouse conditions at 25℃ with a 16 h light period. The virus was purified from infected tobacco as described in a previous report (Gooding G V J, et al., 1967). TMV-infected leaves were mixed with 0.5 mol/L phosphate buffer (1% BME; 0.01 mol/L EDTA-Na, pH 7.4), homogenized in RNase-and DNase-free mortar and filtered by double RNase-and DNase-free gauze. The supernatant was treated with n-butanol (25:1; v:v), mixed, and centrifuged for 30 min at 1, 000 g. Next, the supernatant was treated with PEG6000 at a final concentration of 4% until the mixture was sufficiently dissolved, incubated at 4℃ for 1 h, and centrifuged for 30 min at 10, 000 g. The precipitate was suspended in 0.01 mol/L phosphate buffer (pH 7.4), followed by differential centrifugation for 120 min at 30, 000 g and 5 min at 10, 000 g. Following re-suspension in 0.01 mol/L phosphate buffer (pH 7.4), the precipitate was centrifuged for 10 min at 1, 000 g. The supernatant containing TMV was collected and stored at 4℃ and used as mother liquor in subsequent experiments.
-
In 2012, 28 water samples were collected from different tobacco production areas in the Shaanxi Province of China. All samples were filtered through double gauze, delivered to the laboratory within 24 h, and pretreated by centrifuging for 10 min at 1, 000 g. Supernatants were collected and stored at -80℃ for later use.
-
Gene sequences of nine different TMV isolates were obtained from NCBI (Genbank accession numbers AF395127.1, AF395128.1, AF395129.1, HE818416.1, HE818448.1, HE818453.1, HE818455.1, HE818457.1 and NC-001367.1) and analyzed using the program DNAMAN. Four specific primer sets were designed based on the genome sequences, using the Primer Premier 5.0 program. The primer sequences, expected sizes of amplification products and target genes are listed in Table 1.
Primer Sequence 5'-3' Positions Amplicon size (bp) F1 AGCCTTTTGCGGTGACGATAGT 4490-4511 242 R1 ACTCCTCCAAGTGTTCCCAATC 4710-4731 F2 GCATCGCATCTGTTCAAGGGAC 405-426 271 R2 GTAGCGCAATGGCGTACACTCT 654-675 F3 TGTGTCGGTGTGTATTGTTT 5379-5398 204 R3 CTACTATTTTTTCCCTTTGCG 5563-5582 F4 TAAAGCACAACCCAAGCAA 3977-3995 117 R4 TCACTAAACAACGGGCAAA 4074-4093 Table 1. Primers sequences for detecting TMV
-
RNA was extracted from mother liquor using the Supermo RT Kit (Bioteke, Beijing, China), and cDNA synthesized using the Prime Script RT reagent Kit (TaKRa, Dalian, China)according to manufacturer's instructions. A 25 μL reaction mixture containing 2 μL cDNA, 12.5 μL 2×Premix Ex Taq, 400 nmol/L of each primer and ddH2O was subjected to PCR in the PTC-100 Peltier Thermal Cycler (Bio-Rad, California, USA). The amplification conditions were as follows: denaturation at 94℃ for 3 min, 35 cycles of 95℃ for 30 sec, 51℃ for 30 sec and 72 ℃ for 30 sec, 72℃ for 10 min, followed by incubation at 4℃. Products were detected via electrophoresis on a 2.5% agarose gel, and UV light used to visualize the bands. Amplification products obtained using specific primers were confirmed by purification from agarose gels using a Gel Extraction Kit (Bioteke, Beijing, China)following the manufacturer's instructions. DNA fragments were cloned into the pMD18-T simple vector, transformed into Escherichia coli JM109 (heat shock at 42℃ for 90°s), and sequenced by Sangon Biotech Co., Ltd (Shanghai, China).
Positive plasmids were quantified by measuring the optical density at 260 nm (OD260), and the viral copy number calculated using the following formula:
To evaluate and optimize the sensitivity of real-time PCR, a series of 10-fold serial dilutions of standard plasmid were prepared. The starting concentration of standard plasmid was 1010 viral copies/μL, and the series ranged from 1 to 107 viral copies/μL. Serial dilutions were assayed via real-time PCR in triplicate in eight independent runs. The reaction mixture consisted of 12.5 μL 2×SYBR Green I PCR master mix, 2 μL template cDNA, ddH2O, and 200 nmol/L specific primers. Reactions were conducted using the IQ5 rt-PCR Detection System (Bio-Rad, California, USA). The optimized conditions were as follows: 95℃ for 10 min, 45 cycles each consisting of 15 sec at 95℃ and 30 sec at 51℃ (annealing and extension). The sizes of real-time PCR products were examined on a 2.5% agarose gel. Serially diluted plasmids were used to establish a standard curve for TMV by plotting the threshold cycle value against the viral copy logarithm via linear regression.
-
Tobacco etch virus (TEV), Cucumber mosaic virus (CMV), and Potato virus Y (PVY) are members of the virus family causing serious disease to tobacco, and mixed infection with TMV is common. All three viruses have been detected in water (Zheng Y T, et al., 2000), and therefore selected to determine the specificity of real-time PCR. All tobacco samples infected by the viruses were stored under greenhouse conditions at 25℃ with a 16 h light period.
-
The standard water sample was generated by dissolving 1 μL mother liquor in 10 mL distilled water. The solution was placed in a 15 mL centrifugal tube to which PEG6000 (Sangon Biotech Co., Ltd, Shanghai, China) was vigorously mixed. Following incubation at room temperature for 30 min, the mixture was centrifuged for 30 min at 4℃. The precipitate was re-suspended in 30 μL of 0.01 mol/L Phosphate Buffer Saline (PBS, pH = 7.4) and transferred to a 1.5 mL RNase-and DNase-free centrifuge tube. Virus RNA was extracted from the solution, reverse-transcribed in a total reaction volume of 25 μL, and cDNA used for real-time PCR, according to the manufacturers' procedures. The concentration factors, final PEG6000 concentration and centrifugal force, were further tested for optimization. PEG6000 at final concentrations of 1%, 10%, 20%, 30% and centrifugal force of 3000 g, 4000 g, 5000 g, 6000 g, and 7000 g were subjected to orthogonal test.
To ascertain the sensitivity of the concentration procedures, a series of 10-fold serial dilutions of standard water samples were prepared. The starting concentration in the standard water sample was 107 viral copies/μL, and the dilution series ranged from 1 to 107 viral copies/μL.
-
N. glutinosa resistant to TMV and N. tabacum L.NC89 (NC89) susceptible to TMV were cultivated in a disease-free greenhouse and used for the infectivity tests. N. glutinosa with four or five leaves was selected, and two leaves mechanically inoculated with TMV after concentrating from standard water. Inoculated leaves of all plants were monitored until the appearance of symptoms in the next 14 days.
NC89 was also used to test the infectivity of TMV after concentrating. In this case, all plants were cultivated in a hydroponic system. The original viral concentration was serially diluted 10-fold (from 100 to 104 viral copies/mL) in liquid medium. Five plants were planted in liquid medium with 10-fold dilutions of the original viral concentration, and an additional five plants used as the negative control. After two weeks, the upper leaves of the plants were analyzed using RT-PCR.
-
To determine the optimal method for detection of virus in environmental water, 28 water samples were concentrated and analyzed using the above procedures. Simultaneously, concentrates of the samples were additionally examined using RT-PCR and DAS-ELISA to confirm the results of real-time PCR. For ELISA, antisera were purchased from Neogen Corporation (Shanghai, China), and the procedure performed in keeping with the manufacturer's instructions.
Virus
Water samples
Design and selection of TMV-specific primers
Reverse transcription PCR and real-time PCR
Specificity of real-time PCR
Concentration and detection of TMV in standard water
Infectivity tests
Detection of TMV in environmental water samples
-
Primers (Table 1) and annealing temperatures for real-time PCR were optimized through a series of tests. The results indicate non-specific amplification in reactions with primers F1/R1 and F3/R3 and < 90% amplifi-cation efficiency with F4/R4. Using F2/R2, amplification efficiency reached 96.7%, with no detection of nonspecific products. Primers F2/R2 and annealing temperature of 51℃ were considered optimal and applied for subsequent experiments.
Standard curves of real-time PCR were established using standard plasmid ranging from dilutions of 102 to 107 viral copies/μL. Linear correlation (R2) between the threshold cycle and viral copy logarithm was 0.999 with a slope of -3.403, indicating a reproducible linear response in detection of TMV (Figure 1).

Figure 1. Standard curve of real-time PCR based on a 10-fold serial dilution of standard plasmid. Serially diluted plasmid (102 to 107 viral copies/μL) was used to establish the standard curve for TMV.
The sensitivity of real-time PCR was tested using eight 10-fold serial dilutions of standard plasmid. Upon comparison of the eight independent runs, TMV was detected in all except one dilution sample (1 viral copy/μL). The threshold cycle for dilution was 33.96, similar to the negative control. Accordingly, the detection limit of real-time PCR was set as 10 viral copies/μL (Figure 2).
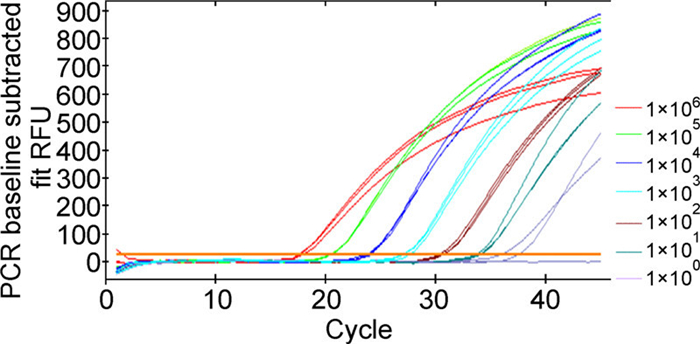
Figure 2. Sensitivity of real-time PCR. The positive plasmid was diluted serially 10-fold (100 to 106 viral copies/μL) with deionized water to evaluate the sensitivity of the assay. RFU in the figure stands for PCR baseline subtracted fit relative fluorescence units; Amplification plots stand for 1×106, 1×105, 1×104, 1×103, 1×102, 1×101, and 1×100 viral copies/μL respectively.
The specificity of real-time PCR for TMV was investigated with three viruses (CMV, TEV and PVY). No fluorescence was detected for the three viruses and negative control, confirming the high specificity of the assay.
-
Temperature, pH, turbidity, organic material concentration and centrifugal force are the common factors affecting the concentration of viruses in water (Colombet J, et al., 2007). PEG is able to promote precipitation under various conditions (Colombet J, et al., 2007). Therefore, among the concentration factors, final PEG6000 concentration and centrifugal force were specifically optimized. Final PEG6000 concentrations of 1%, 10%, 20%, 30% and centrifugal forces of 3000, 4000, 5000, 6000 and 7000 g were analyzed via orthogonal test. Concentration of virus, RT-PCR and real-time PCR analyses were performed as described previously. Measurement of recovery efficiency, using the equation E (%) = (viral copy of recovered TMV/viral copy of initial seeded TMV) ×100, was used to assess different treatments. To obtain purified TMV that can be directly detected via real-time PCR, PEG was removed using KCl (Colombet J, et al., 2007). Comparison of the recovery efficiencies of different treatments revealed 20% final PEG6000 concentration and 6000 g centrifugal force as the optimal conditions, leading to 90.95% genome recovery (Table 2).
Centrifugal Force (g) PEG6000 concentration (%) 1 10 20 30 3000 0.70%a
(0.05%)b13.10%
(0.05%)11.63%
(0.06%)8.89%
(0.08%)4000 4.07%
(0.05%)16.66%
(0.05%)21.02%
(0.06%)17.50%
(0.08%)5000 8.66%
(0.12%)24.34%
(0.20%)50.36%
(0.16%)38.52%
(0.05%)6000 9.89%
(0.03%)44.79%
(0.11%)90.95%
(0.16%)62.02%
(0.05%)7000 8.68%
(0.26%)32.82%
(0.22%)41.88%
(0.13%)11.40%
(0.12%)Orthogonal design tests were carried out at room temperature, and the pH of standard water was ~7.2.aWithin columns indicates recovery efficiency, calculated according to the equation E (%) = (viral copy of recovered TMV/viral copy of initial seeded TMV) ×100; bwithin columns signifies standard error. Table 2. Orthogonal design tests for method optimization
The sensitivity of the concentrated procedures was tested with eight 10-fold serial dilutions of standard water. Upon comparison of the eight independent runs, three dilutions (ranging from 102 to 1 viral copy/μL) were negative for TMV. The detection limit of the concentration procedures was therefore set as 103 viral copies/μL (Figure 3).

Figure 3. Sensitivity of the concentration method. A series of 10-fold serial dilutions of the standard water sample were prepared (amplification plot a-h). RFU in the figure stands for PCR baseline subtracted fit relative fluorescence units; The starting concentration of the standard water sample was 107 viral copies/μL (amplification plot a), and amplification plots stand for 1×107, 1×106, 1×105, 1×104, 1×103 viral copies/μL respectively.
-
The infectivity of TMV after concentrating was tested using N. glutinosa and NC89 strains. Following inoculation of N. glutinosa with TMV, necrotic lesions developed after 4-6 days on leaves while the control PBS-treated leaves displayed no symptoms (Figure 4). This finding clearly indicates that TMV maintains infectivity after concentration with PEG.

Figure 4. Infectivity of TMV in N. glutinosa after concentrating. Upon inoculation of N. glutinosa with TMV, necrotic lesions developed after 4-6 days. A: N. glutinosa leaf infected with TMV concentrated from the mother liquid via mechanical inoculation. B: N. glutinosa leaf inoculated with 0.01 mol/L phosphate buffered saline (PBS, pH 7.4).
In NC89 cultivated using the hydroponic system, necrotic symptoms on the leaves of all plants were not observed, even after two weeks. However, infectivity of TMV after concentrating showed no signs of degradation, and the virus could re-infect plants through the roots. Among the five plants cultivated in liquid with TMV, three tested positive (Figure 5). The two remaining plants cultivated in liquid medium with 101 and 100 viral copies/mL tested negative for TMV.

Figure 5. Infection test for detecting the activity of TMV in concentrates. All plants were cultivated using a hydroponic system. The F2/R2 primer set was used to detect TMV infection, and the size of PCR product was 271 bp. The original viral concentrate was serially diluted 10-fold (100 to 104 viral copies/mL) in liquid medium. M: DM2000; Lanes 1-5: 1×104, 1×103, 1×102, 1×101, 1×100 viral copies/mL; Lane 6: Positive control; Lane 7: Healthy tobacco (NC89) control.
-
To assess the applicability of the concentration procedures used for detecting virus in environmental samples, 28 samples were examined before and after virus concentration. Before concentration, no samples displayed positivity for TMV with DAS-ELISA. After concentration, real-time PCR, RT-PCR and DAS-ELISA were employed to compare efficiency of virus detection. The same result was obtained for 5 samples with all three methods, 7 samples tested positive for TMV using real-time PCR and RT-PCR but were negative with DASELISA, while 5 samples were positive for TMV using real-time PCR but negative with DAS-ELISA and RTPCR. Additionally, 11 samples were negative for TMV with all three methods (Table 3).
Sample Real-time PCR RT-PCR ELISA Location Number Positive sample Positive sample Positive sample Long County 8 5 4 1 Xunyang County 9 7 5 2 Fu County 6 3 2 1 Nanzheng County 5 2 1 1 Total 28 17 12 5 Table 3. Clinical detection results with real-time PCR, RT-PCR and ELISA
Optimization of TMV-specific real-time PCR
Optimization of the method for concentrating TMV
Infectivity tests
Detection of field samples with real-time PCR
-
TMV, one of the most serious diseases in tobacco, has been found in almost all tobacco fields in China (Chen R, et al., 1997). In addition to aphids, seeds and direct contact, water has been identified as another means to spread TMV (Zheng Y T, et al., 2000). A rapid, specific and sensitive detection method is therefore necessary for monitoring the water supply used for crop irrigation. In this study, PEG6000 was used as the precipitating agent to concentrate TMV in standard water, and real-time PCR subsequently employed to detect and quantify TMV in water. The optimized method was proved to be effective in detecting TMV within water.
PEG was identified as an excellent precipitation agent for concentrating TMV from water in our experiments. Viruses exist in natural or tap water at significantly low levels (Li J W, et al., 1998). A suitable concentration procedure is therefore required for TMV detection. TMV could not be detected in all water samples in our DASELISA experiments. After concentrating using a PEGbased procedure, 5 samples tested positive for TMV using DAS-ELISA, supporting the efficacy of PEG in concentrating TMV from water to a level above the sensitivity threshold of this method.
The concentration of PEG was distinguished as an important influencing factor in the virus concentration method. In interactions between protein and PEG, the effectiveness of PEG is enhanced with increasing concentrations in water samples. Owing to non-specific interactions, minor components, proteins and viruses are separated constantly from water at increasing PEG concentrations. In the current study, recovery efficiency increased significantly in proportion to a range of final PEG 6000 concentrations from 1% to 20%. In contrast, from 20% to 30% PEG6000, recovery efficiency was markedly decreased. Maximal recovery efficiency was observed at a final PEG6000 concentration of 20% (Table 2), which was subsequently used for concentrating virus.
The viability of TMV was further ascertained. In infectivity tests, hypersensitive response (HR) was observed in the leaves of N. glutinosa mechanically inoculated with TMV. Additionally, TMV was able to re-infect NC89 through the roots (Figure 3). These results confirm that TMV is not destroyed during the concentration process, and its viability is not degraded in water. However, TMV concentration in water was identified as an important factor for reinfection efficiency. Two plants cultivated in liquid medium with 101 and 100 viral copies/mL tested negative for TMV (Figure 3). TMV infects plants though their roots in water. At extremely low concentrations of TMV in water (101 viral copies/mL), the probability of TMV reaching the plant root is reduced, resulting in failure of reinfection. In addition, TMV, a single RNA virus, may be degraded easily in vitro, which may be one of the reasons underlying its failure to reinfect plants at low concentrations.
Water samples collected from different tobacco production areas in Shaanxi Province were tested for virus detection with real-time PCR, RT-PCR and DAS-ELISA. Overall, 17 samples tested positive for TMV with real-time PCR, 12 with RT-PCR and 5 with DAS-ELISA (Table 3). These findings reflect the different sensitivities of the three procedures, and are consistent with earlier reports that real-time PCR is more sensitive than the other methods in the detection of low-level viruses (Agindotan B O, et al., 2007).
In conclusion, our findings demonstrate the potential application of real-time PCR, combined with PEG6000-based concentration, to effectively detect TMV from samples of irrigation water. Compared with other methods, this technique is easier to perform, cost-effective and more efficient. Therefore, the protocol developed in this study presents a reliable and rapid tool to screen for the existence of TMV in environmental water.
-
We thank Guo Ruizhen for editing the manuscript and Tao Ye of NWSUAF for corrections and suggestions. The study was supported by the Key Technology Program of China National Tobacco Corporation (110200902046), 111 Project from the Education Ministry of China, (No. B07049) and the National High-tech R & D Program of China (no. 2012AA101504).
-
All authors declare that they have no competing interests. And no human and animals were used in the whole experiments.
-
Wei Chen did the experiments, carried out data analysis and drafted the manuscript. Wenting Liu, Honghong Jiao, and Huawei Zhang participated in the experiments. Julong Cheng collected samples. Yufeng Wu conceived and supervised the study.

 DownLoad:
DownLoad: