









HTML
-
Monkey B virus (Macacine herpesvirus 1; BV) is a macaque α-herpesvirus closely related to the human herpes simplex viruses (HSV1 & HSV2)(Elmore D, et al., 2008; Huff J L, et al., 2003). In its natural macaque host BV behaves very much like HSV does in humans. Most macaques acquire BV either orally as infants or as adults via sexual transmission. Primary BV infections are usually asymptomatic, the virus establishes latency in sensory neurons, and latent virus can reactivate and be shed in oral or genital secretions in response to stress. However, when transmitted to other primate species including humans, BV usually invades the central nervous system (CNS) resulting in an ascending encephalomyelitis and death. Although only about 50 cases of zoonotic BV infection have been documented, approximately 80% of untreated cases are fatal and survivors frequently experience subsequent neurological deterioration. Consequently, BV is the primary zoonotic concern for research, veterinary, and animal caretakers working with macaques.
Nothing is known regarding viral genes that facilitate the cross-species neurovirulence of BV. Since BV and HSV are closely related, it has been widely assumed that what is true for HSV applies equally to BV. However, as research on BV progresses it becomes more apparent that this is not always true. For example, while the γ34.5 gene of HSV is a major determinant of neurovirulence (Chou J, et al., 1990; Jing X, et al., 2004; MacLean A R, et al., 1991), this is the only HSV gene that BV lacks a homologue of (Ohsawa K, et al., 2014; Perelygina L, et al., 2003).
Another gene shown to affect neurovirulence of HSV in mice is UL41. This gene encodes the virion host shutoff (vhs) protein, and homologues of the UL41 gene are present in most α-herpesviruses that have been sequenced (Smiley J R, et al., 2001). The vhs proteins of HSV and related α-herpesviruses have sequence homology with some mammalian RNases, and the HSV vhs protein has been shown to have RNase activity (Esclatine A, et al., 2004a; Everly D N, et al., 2002; Zelus B D, et al., 1996). The vhs protein is a component of the virion tegument, and so is deposited into the cytoplasm of the host cell when the viral envelope fuses with the host cell plasma membrane. The vhs protein degrades cellular mRNAs via its RNase activity, contributing to decreased synthesis of host proteins in the infected cell (Esclatine A, et al., 2004a; Karr B M, 1999; Taddeo B, et al., 2006) and to enhanced expression of viral late genes (Dauber B, et al., 2011; Taddeo B, et al., 2013). However, this mRNA degradation is selective in that not all mRNAs are equally affected (Esclatine A, et al., 2004a; Esclatine A, et al., 2004b). It has also been shown that vhs is able to alter translation of bicistronic mRNAs by suppressing expression of the 5’ cistron and activating expression of the 3’ cistron (Saffran H A, et al., 2010), an observation of significance given the polycistronic nature of a number of herpesvirus mRNAs.
Since the interferon-β(IFN-β) response is a major facet of the host innate immune response to viral infections, the ability to prevent or delay this response gives the virus a distinct advantage in gaining a foothold in the host (Mossman K L, 2005; Takeuchi O, et al., 2007). Degradation of host cell mRNAs is one mechanism by which α-herpesviruses prevent the infected cell from mounting an IFN-β response (Duerst R J, et al., 2004; Pasieka T J, et al., 2008; Suzutani T, et al., 2000). Deletion or truncation of the UL41 gene of HSV or the baboon α-herpesvirus HVP2 allows infected mouse cells to mount an efficient IFN-β response (Duerst R J, et al., 2004; Rogers K M, et al., 2009). Consistent with this, deletion of the UL41 gene has been shown to impair pathogenicity of the virus (Duerst R J, et al., 2004; Korom M, et al., 2008; Rogers K M, et al., 2009; Smith T J, et al., 2002; Strelow L I, et al., 1995).
This study was undertaken to determine if the vhs protein of BV is a critical factor in supporting viral replication and invasion of the CNS, and possibly a factor in the extreme neurovirulence of BV.
-
BV strain E90-136 was originally isolated from a young cynomolgus macaque that developed a fatal systemic infection (Simon M A, et al., 1993) and was used as the parental wildtype (WT) virus for all experiments. Virus stocks were prepared and quantitated in Vero cells, and experiments were performed in either Vero cells or primary mouse dermal fibroblast (PMDF) cultures as previously described (Hilliard J K, et al., 1987; Rogers K M, et al., 2006; Rogers K M, et al., 2007). All work with infectious BV was conducted using procedures and facilities approved by the OSU Biosafety Committee and in compliance with the OSU Select Agent Program as approved by the US Centers for Disease Control and Prevention.
-
The UL41 open reading frame (ORF) was amplified by PCR from purified viral DNA and cloned into pUC19. Three amino acid (AA) mutations (191E→Q, 193D→N & 194D→N; Figure 1) were introduced into the vhs RNase active site by PCR amplification of the cloned UL41 ORF using overlapping primers that spanned the mutation sites and incorporated a unique Sph 1 restriction site. Amplified products were then digested with Sph 1, ligated, and transfected into DH5α cells. The integrity of the mutated UL41 ORF was confirmed by sequencing.

Figure 1. Location of mutations introduced into the vhs protein. Alignment of the predicted amino acid sequences of vhs proteins of HSV1, HSV2, HVP2, the attenuated reference BV strain (E2490; BVrh) (Hull, 1971) and BV field isolate strain E90-136 (BVcy) are shown. The RNase active site is indicated below and mutations introduced to make the E90-136 41ΔA mutant are shown above the sequences.
Mutant viruses were generated as previously described for HVP2 (Rogers K M, et al., 2009). Briefly, the UL41 ORF was deleted from the wild-type (WT) BV genome and replaced with a green fluorescent protein (GFP) expression cassette (Black D H, et al., 2002). This UL41 ORF deletion virus was designated Δ41.
To generate a virus carrying the RNase active site mutations, Vero cells were transfected with a construct consisting of the mutated UL41 ORF plus flanking sequences, the transfected culture infected 24-48 hrs later with the Δ41 mutant, and GFP-negative plaques picked and purified by multiple rounds of limiting dilution as described (Rogers K M, et al., 2009). The UL41 RNase mutant was designated Δ41A. The same procedure was followed using the WT UL41 ORF to replace the GFP expression cassette, and the revertant virus designated Δ41R. The integrity of all mutants was confirmed by PCR/sequencing.
-
All animal experiments were reviewed and approved by the OSU Institutional Animal Care and Use Committee and were conducted in compliance with the OSU Select Agent Program. For determination of 50% lethal dose (LD50) and 50% infectious dose (ID50) values, groups of 8–13 Balb/c mice (10–12 g) were inoculated with 10-fold dilutions of virus (106–101 PFU) by scarification of the skin on the left flank as described (Rogers K M, et al., 2006). For all other experiments an inoculation dose of 105 PFU was used (~10 LD50). Mice were observed twice daily for clinical signs of disease. Any mouse exhibiting signs of severe CNS infection (tremors, immobility, bilateral hind limb paralysis) or autonomic nervous system (ANS) dysfunction (severe dehydration, abdominal distension/bloating, diarrhea, urinary incontinence) were humanely euthanized. Mice surviving to 14 days PI were bled and euthanized. Tissues were collected at necropsy and processed for histopathological examination as described (Ritchey J W, et al., 2002).
-
Total RNA was extracted from Vero cells infected at an MOI = 5 PFU/cell at 4 hrs PI using an RNAeasy kit (QIAGEN). RNA was run on formamide agarose gels (10 µg/well) and transferred to nitrocellulose for hybridization. Probes for northern blot were prepared by nick translation of the cloned Vero cell actin gene and hybridized to blots as described (Rogers et al., 2009). Autoradiographs were quantitated using the NIH Image program (http://rsb.info.nih.gov/nih-image/). All infected cell values were adjusted based on the 28S RNA as a loading control and subsequently normalized to the mock infected cell sample.
-
IFN-β was quantitated using a commercial ELISA according to the manufacturer’s instructions (PBL Interferon Source, Piscataway, NJ) as previously described (Rogers et al., 2009). Details of ELISA for detection of anti-BV IgG, SDS-PAGE, and western blot analysis of infected cell proteins have also been described (Hilliard et al,. 1987; Ritchey et al,. 2002). Antiserum raised to the HSV1 vhs protein (Strelow and Leib, 1995) was kindly supplied by Dr. David Leib (Dartmouth College, Hanover, NH) and was used at a dilution of 1:1000.
-
ID50 and LD50 values were calculated by probit regression with PROC PROBIT in PC SAS Version 9.1 (SAS Institute, Cary, NC, USA), and values were compared by methods developed for effective dosages. Virus titers in experimental samples were compared by analysis of variance procedures with PROC MIXED in PC SAS. Virus titers were transformed using log base 10 function prior to analysis. Significant differences were determined by protected pair-wise t tests with a SLICE and DIFF options in an LSMEANS statement.
Viruses and cell culture
Construction of UL41 mutants
Mouse inoculations
Northern blot
Immunoassays
Statistical analyses
-
The UL41 gene/vhs protein of HSV is dispensable for virus replication in Vero cells, and consistent with this WT BV, the revertant (Δ41R) and both UL41 mutants (Δ41 & Δ41A) all replicated with equal efficiency in Vero cells; the amount and temporal appearance of progeny virus produced by the revertant and the two mutants did not differ significantly from the WT virus (not shown). While all viruses were also able to form foci of infection in primary mouse cells by 24 hrs PI, foci of Δ41 and es-pecially Δ41A were smaller than those of WT or Δ41R (Figure 2A). By 48 hrs PI both WT and Δ41R plaques had dramatically expanded and by 72 hrs PI involved the entire cell monolayer. In contrast, while Δ41 foci slightly enlarged in size after 24 hrs, but they never progressed to involve the entire cell monolayer while Δ41A foci did not expand in size past 24 hrs PI and became overgrown by uninfected cells. Quantitative assessment of replication in PMDF cells showed a maximal increase of infectious WT and 41R by 16 hrs PI, after which time titers slowly decreased (Figure 2B). In contrast, there was little to no increase in infectious Δ41 or Δ41A over input virus levels, and past 24 hrs PI titers of these two mutants dropped significantly, indicating that mutation or deletion of vhs affects the ability of BV to replicate in primary mouse cells.
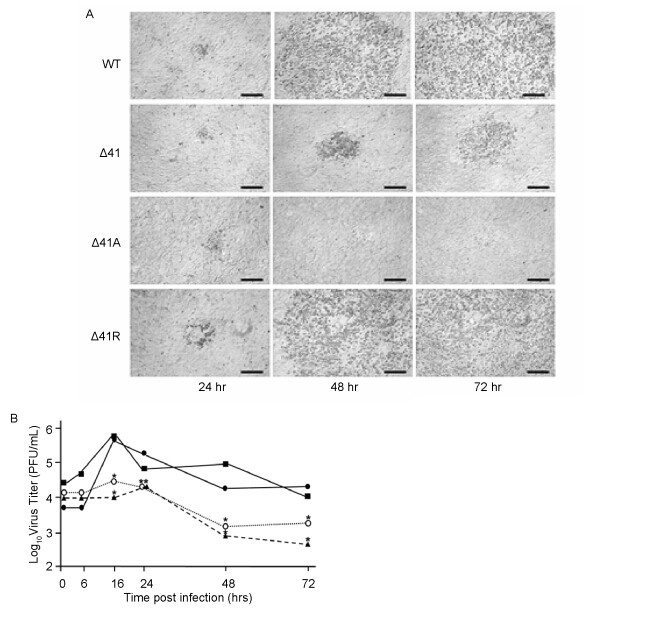
Figure 2. Replication of BV UL41 mutants in primary murine cells. PMDF cultures were infected at an MOI = 0.03 PFU/cell and plaque development observed (A). Representative foci of infection were photographed at 24, 48 & 72 hrs PI. Bars = 200 µm. Quantitation of viral replication in PMDF cultures (B) was assessed by infecting at an MOI = 0.01 PFU/cell, harvesting cells at the indicated time points, and quantitating infectious virus by plaque assay on Vero cells. Viruses were WT (■——■), Δ41R (●——●), Δ41 (○-----○) and Δ41A (▲−−−−▲). Values at each time point that were significantly different from those of the WT virus (p < 0.05) are indicated by asterisks.
Western blot confirmed that a vhs polypeptide was synthesized in WT, Δ41R and Δ41A infected Vero cells, but not in Δ41 infected cells (Figure 3A). A number of proteins synthesized during the 2-8 hr PI labeling period in mock infected cells were not evident by SDS-PAGE in cells infected with either WT or Δ41R (Figure 3B), confirming that these two viruses efficiently suppress synthesis of host cell proteins. In contrast, the Δ41 and Δ41A mutants did not suppress host cell protein synthesis following infection. Consistent with this, both WT and Δ41R efficiently degraded β-actin mRNA while the Δ41 and Δ41A mutants did not (Figure 3C). In fact, β-actin mRNA levels were somewhat higher in Δ41 and Δ41A infected cells than in mock infected cells (~1.5-fold and ~2.5-fold, resp.). Consistent with its almost complete inability to replicate in PMDF cells (Figure 2A and 2B), it is also apparent in Figure 3A that the Δ41A mutant produced less virus-specific proteins than the other viruses.
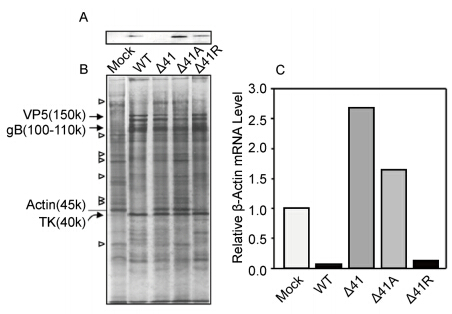
Figure 3. Host shutoff function of vhs mutants. PMDF cultures were infected with virus (MOI = 5 PFU/cell) and labeled with 35S-Met/Cys from 2-8 hrs PI, harvested at 8 hrs PI, and proteins separated by SDS-PAGE. The vhs protein was detected by western blot using a polyclonal antiserum to recombinant HSV1 vhs (A). An autoradiograph of the blot is shown in (B). Host cell proteins synthesized in cells infected with the vhs mutants but not detectable in WT or Δ41R infected cells are indicated by open arrowheads. As molecular weight standards, several viral proteins are identified by solid arrowheads. Degradation of β-actin mRNA following infection was assessed by northern blot (C). Total RNA was extracted from mock infected or virus infected Vero cells at 4 hrs PI, run on a gel, blotted to a membrane, and hybridized with a β-actin probe. Quantitation of a representative blot is shown where the amount of β-actin mRNA was adjusted based on a 28S RNA loading control and normalized to levels present in mock infected cells.
An important innate antiviral response of cells to infection is induction of an IFN-β response. WT virus was able to efficiently suppress this response (Figure 4). At a high MOI (4 PFU/cell) the Δ41R revertant com-pletely suppressed the IFN-β response, but low levels of IFN-β were detected at a lower MOI (0.4 PFU/cell). In additional experiments no IFN-β response was elicited by Δ41R infection at either MOI (not shown). PMDF cultures infected with Δ41 or Δ41A consistently produced high levels of IFN-β at all MOIs tested, and Δ41A infected cultures produced significantly higher levels of IFN-β than Δ41 infected cultures at low MOI. The higher level of IFN-β produced by Δ41A relative to Δ41 infected cells is consistent with the inability of Δ41A to form plaques and Δ41 to only form small plaques in primary mouse cells.
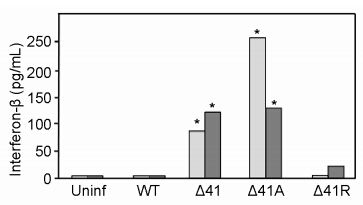
Figure 4. vhs dependent suppression of the host IFN-β response. PMDF cultures were infected at an MOI = 0.4 (light gray) or 4.0 (dark gray) PFU/cell. At 24 hrs PI, medium was collected and IFN-β levels quantitated by ELISA. The results shown are for one of three experimental repeats. Values that differed significantly (p < 0.05) from WT virus IFN-β levels are indicated by asterisks.
-
Neuropathogenicity of the BV UL41 mutants, both their ability to invade the CNS and produce neurological disease following inoculation at a peripheral site, was tested using a mouse model. LD50 values for WT (104.3 PFU) and Δ41R (104.1 PFU) were not significantly different from one another (p < 0.05) and were similar to values previously reported for other neurovirulent BV isolates (Ritchey J W, et al., 2002). All mice that survived inoculation with WT or Δ41R lacked serum anti-BV IgG, indicating that they had not experienced an active infection, making the ID50 and LD50 values the same for these viruses; if BV was able to establish infection, it was always lethal. LD50 values for Δ41 (105.3 PFU) and Δ41A (105.1 PFU) were significantly higher (p < 0.05) than the LD50 for WT or Δ41R. A number of Δ41 and Δ41A infected mice that survived past 11 DPI had serum antiBV IgG, resulting in ID50 values of 104.6 and 104.9 PFU for Δ41 and Δ41A, respectively. Thus, while the Δ41 and Δ41A mutants can establish active infections nearly as efficiently as the WT virus, not all infections with these two mutants were lethal.
The pathogenesis of vhs mutants was compared to that of WT BV. Mice were infected with 105 PFU of virus by skin scarification on the hind flank and observed for clinical signs of infection for 14 days. As summarized in Figure 5, the temporal pattern of disease was indistinguishable for WT and Δ41R. At 4 DPI vesicular lesions appeared on the skin at the site of inoculation, followed by rapid and sequential development of abnormal adduction of the ipsilateral hind leg when lifted by the tail (a reflex arc involving interneurons in the spinal cord), paresis followed by paralysis of the ipsilateral hind leg (which sometimes progressed to involve the contralateral limb as well), reluctance to move, tremors, and death/euthanasia. Skin lesions worsened and/or spread with time in some mice, but this was not consistently observed. Histological examination of the CNS and ANS of WT and Δ41R infected mice at the time of euthanasia (5-6 DPI) revealed lesions characterized primarily by abundant cell debris (indicative of necrosis), spongiosis, and evidence of viral replication (intranuclear inclusion bodies and cell destruction). Although there was lymphocytic infiltration, this was primarily restricted to the dorsal meninges and outermost dorsal funiculus; inflammation was less conspicuous than lesions of cellular destruction.

Figure 5. Temporal appearance of clinical signs of disease in infected mice. Groups of mice (n = 11-13) were infected with 105 PFU of virus on the left flank by skin scarification (Rogers et al., 2009). The cumulative number of mice displaying each symptom at each day PI is shown. Clinical signs followed were the appearance of vesicular skin lesions at the site of inoculation (A), abnormal abduction of the left leg/foot when lifted by the tail (B), paresis (C) and paralysis (D) of the left hind leg, and death/euthanasia (E). Viruses were WT (■——■), Δ41R (●—●), Δ41 (○-----○) and Δ41A (▲−−−−▲).
Relative to the rapid course of disease progression seen in WT infected mice, mice infected with the two vhs mutants displayed a progressively delayed time course of infection (Figure 5). Clinical signs of infection developed in the same sequential progression as in WT infected mice, but the time of first appearance of these signs was delayed in Δ41 and Δ41A infected mice. Appearance of skin lesions and abnormal adduction of the ipsilateral hind leg was delayed by an average of 1 day, paresis of the ipsilateral hind leg by 2 days, paralysis by 3 days, and death/euthanasia by 4 days. This progressive temporal delay suggests that for these two mutants spread of the infection is delayed at all points in the pathogenic process.
While all Δ41 infected mice that developed clinical signs of CNS infection progressed to require euthanasia, only 4/7 Δ41A infected mice with clinical signs of CNS infection required euthanasia, and these 4 mice were euthanized due to symptoms associated with ANS dysfunction (urinary retention/incontinence, severe distension of the cecum/colon, severe dehydration) rather than for CNS symptoms. Skin lesions completely healed in Δ41A infected mice and, unlike Δ41 infected mice, secondary skin lesions did not develop. In addition, 2/3 Δ41A infected mice that developed hind limb paralysis but were not euthanized for signs of ANS dysfunction recovered some use of their paralyzed leg by termination of the experiment (16 DPI for these three mice). These results are consistent with plaque formation and IFN-β expression in infected primary mouse cells in that the presence of a non-functional vhs protein appears to be more detrimental to the virus than the complete lack of the vhs protein.
Histopathologically, Δ41 and Δ41A infected mice had lesions in the skin, spinal cord, dorsal root ganglia, sympathetic ganglia, and muscular tunics of the urinary bladder and cecum/colon. Skin samples from the site of inoculation of Δ41A infected mice either had no significant lesions (lesions never developed or were completely resolved at euthanasia) or mild to moderate hyperplastic lymphohistiocytic dermatitis, while skin lesions from Δ41 infected mice exhibited ulceration, crusting, adjacent epithelial hyperplasia, and more intense dermatitis accompanied by granulation. CNS and ANS lesions in Δ41 and Δ41A infected mice were wholly inflammatory (lymphocytic) without necrosis of neurons or ganglion cells and without detectable viral inclusion bodies that were so conspicuous in mice infected with WT or Δ41R. Perhaps due to the temporally prolonged disease course, inflammation characterized by lymphocytic perivascular cuffing restricted to the caudoventral brainstem regions was also observed in Δ41 and Δ41A infected mice.
To directly compare the neuropathogenesis of WT and UL41 mutant viruses, a second experiment was performed in which mice were inoculated with 105 PFU, euthanized at 5 DPI, and lesions in the skin (site of inoculation), brain, and lumbar spinal cord were compared. Skin lesions in mice infected with WT, Δ41R and Δ41 all exhibited severe dermatitis, epidermal necrosis, vesicle formation with syncytia, and conspicuous intranuclear inclusion bodies (Figure 6A and 6B) while skin lesions in Δ41A infected mice exhibited primarily ulcerative dermatitis without vesicles or syncytia, and few if any viral inclusion bodies (Figure 6C). The character of lesions in the lumbar spinal cord was similar in mice infected with all viruses, being characterized by meningomyelitis (lymphocytic, neutrophilic) with intranuclear inclusion bodies. The main difference between spinal cord lesions in WT or Δ41R versus Δ41 or Δ41A infected mice was the anatomic distribution and extent of the lesions. Spinal cord lesions in mice infected with both mutants were confined to the ipsilateral dorsal funiculus (Figure 6E and 6F) and were mainly inflammatory with little evidence of tissue destruction. In contrast, spinal cord lesions in mice infected with WT or Δ41R extended into lateral and ventral funiculi and the ventral horn, and exhibited conspicuous necrosis and scattered cellular debris resulting from tissue destruction that regionally obscured cord morphology (Figure 6D). At 5 DPI no lesions were evident in the brainstem of any mice. These results demonstrate that while the Δ41 and Δ41A mutants were neuroinvasive, they produce less severe and more localized lesions than the WT virus.

Figure 6. Comparison of skin and spinal cord lesions at 5 DPI. Skin lesions were qualitatively similar across mice consisting of dermatitis, ulcers and serocellular crusts overlying exposed dermal tissue. WT and Δ41 infected mice exhibited conspicuous syncytial cell formation (A & B, arrows) and intranuclear herpetic inclusion bodies (A & B, arrowheads). Syncytia and inclusion bodies were not features of skin lesions in Δ41A infected mice (C). Spinal cord lesions in mice infected with Δ41 and Δ41A were nearly identical consisting of lymphocytic, neutrophilic meningoencephalitis (E & F, arrows) with occasional intranuclear herpetic inclusion bodies (E & F, arrowheads). Inflammatory lesions and inclusion bodies were restricted to the ipsilateral dorsal funiculus and grey horn. Mice infected with WT BV exhibited inflammation similar to the intensity seen in mutants (D, arrows) and inclusion bodies (D, left arrowhead). In addition, cellular debris (D, middle arrowhead) and neuronal necrosis (D, right arrowhead) was present that extended into the ventral grey horn and the lateral and ventral funiculi. All haemotoxylin & eosin stain; Panel A-C, Bar = 70μm; Panel D-F, Bar = 100 μm.
In vitro characterization of UL41 mutants
In vivo characterization of UL41 mutants
-
The BV vhs protein shares a number of characteristics with the vhs proteins of HSV1, HSV2 and the baboon virus Papiine herpesvirus 2 (HVP2): vhs is dispensable for replication in Vero cells, has RNase activity, and shuts down synthesis of host cell proteins early in infection. The RNase activity of BV vhs is essential for suppression of the cell protein synthesis, mRNA degradation, and suppression of the IFN-β response. Degradation of cellular mRNAs with concomitant suppression of host protein synthesis and the IFN-β response appears to be a conserved and important step in diverting the biochemical machinery of the cell towards replication of α-herpesviruses.
Although deletion or mutation of the UL41 gene does not affect the ability of BV to replicate in Vero cells, replication in primary mouse cells is significantly impaired suggesting that the IFN-β response is likely responsible at least in part for suppressing replication in primary mouse cells. While there was a decreased replicative efficiency of HSV2 UL41 mutants relative to WT virus in primary mouse cells at 24 hrs PI, this difference disappeared by 72 hrs PI rather than increasing as occurs with BV mutants (Korom M, et al., 2008). Despite the extreme neurovirulence of BV strain E90-136 in mice, even the WT virus is unable to sustain high-level replication in primary mouse cells. Mutation or deletion of the vhs protein further reduces the ability of BV to replicate in primary mouse cells to the point that little if any viral replication occurred. It appears that mutation/deletion of the vhs protein allowing an IFNβ response on top of the already poor replication of BV in primary mouse cells accounts for the differences observed between BV and HSV2 vhs mutants in primary mouse cells.
The inability of WT BV to sustain replication in primary mouse cells seems inconsistent with the lethality of BV in mice. Despite inefficient replication in primary mouse fibroblasts in vitro, in vivo replication of WT BV in the skin appears to be a very efficient process since viral inclusion bodies (aggregations of progeny virion nucleocapsids within infected cell nucleii) and syncytia are readily evident in skin lesions. One possible explanation for the apparent discrepancy between in vitro and in vivo results is that in vivo BV is able to efficiently suppress the IFN-β response early in the infection, thereby allowing viral replication in the skin and producing sufficient levels of progeny virus to invade sensory nerve endings in the dermis. Once within the nervous system, the virus would no longer need to replicate efficiently in the skin, obviating the need for sustained replication in skin cells. Although the host can mount an IFN-β response in the CNS as well as in the skin (Paul S, et al., 2007), once within the CNS BV may essentially continue to outpace efforts of the host to control the infection by a combination of impeding host innate defense responses, rapidity of viral replication, and concomitant tissue destruction.
In several respects the Δ41A mutant, which produces a vhs protein that lacks RNase activity, was more severely attenuated than the Δ41 mutant that does not produce any vhs protein: Δ41A could not form plaques in primary mouse cells, allowed production of higher levels of INF-β, did not produce severe or secondary skin lesions in mice, CNS symptoms did not always correlate with lethality, and some Δ41A infected mice recovered use of paralyzed limbs. Thus, the presence in infected cells of a non-functional vhs protein is more debilitating to the virus than not having a vhs protein at all. This could be the result of the non-functional vhs protein interacting with elements of the cellular mRNA degradation system and/or VP16, thereby inhibiting any RNA degradation (Barzilai A, et al., 2006; Taddeo B, et al., 2007); the lack of any vhs protein would not compete a cellular component of the mRNA degradation system, and so permit any residual activity to occur after infection. A non-functional vhs protein could also interact with VP16, and in doing so prevent late viral mRNAs from being protected from degradation (Shu M, et al., 2013).
BV vhs mutants displayed one unique characteristic not observed for HSV2: the accumulation of host cell β-actin mRNA over levels present in uninfected cells. This was observed in every repetition of this experiment, and so appears to be a real phenomenon. While the presence of a vhs protein lacking RNase activity (Δ41A) did result in a slight increase in β-actin mRNA levels over those observed in mock infected cells (1.5-2-fold), the complete absence of a vhs protein resulted in β-actin mRNA levels 2-3 times greater than that in mock infected cells. Such an increase in host mRNA levels was not observed with HSV2 UL41 deletion mutants by Korom et al.(2008) who assessed GAPDH mRNA levels. One possible explanation for this apparent discrepancy is that post infection mRNA degradation may not equally affect all cellular mRNAs. Although degradation of β-actin mRNA was not specifically assessed, selectivity of vhsmediated degradation of mRNAs has been reported (Esclatine A, et al., 2004a; Esclatine A, et al., 2004b). Another possibility relates to the different nature of the BV and HSV2 UL41 deletion mutants. While the entire UL41 ORF was deleted in the BV Δ41 mutant, the HSV2 UL41 null mutant has an in-frame deletion in the UL41 ORF that leaves intact the N-terminal 97 and C-terminal 82 codons of the vhs gene (Smith T J, et al., 2002). Although not detected with anti-vhs antiserum, a 179 AA polypeptide could be produced, and the N- and/or C-terminal vhs sequences constituting it could conceivably function to preclude accumulation of cellular mRNA via interaction with the viral VP16 and/ or VP22 proteins (Smibert C A, et al., 1994; Taddeo B, et al., 2007) or cellular translation initiation factors (Feng P, et al., 2005; Page H G, et al., 2010). The explanation for and significance of β-actin mRNA accumulation in BV infected cells when no vhs protein is present remains an open question.
In summary, mice infected with BV exhibit a fulminant disease course characterized by viral replication in the skin and rapid invasion of the spinal cord accompanied by CNS tissue destruction that outpaces the development of the host inflammatory response. In contrast, mice infected with BV vhs mutants exhibit a delayed clinical course of disease with spinal cord lesions restricted to the ipsilateral dorsal funiculus that are dominated by the host inflammatory response rather than virus-mediated tissue destruction. The vhs mutants are restricted from significant ventral or lateral spread within the spinal cord, yet can move up dorsal ascending tracts to the brainstem and outwards to autonomic ganglia. Although some mice infected with BV vhs mutants survived and others clinically improved, euthanasia of mice infected with the vhs mutants was related to loss of function (spinal cord: paresis/paralysis, autonomic ganglia: bloating/ incontinence) secondary to tissue destruction by the host inflammatory response as reported for HSV1 (Lundberg P, et al., 2008). Although the results of this study clearly show a difference in the clinicopathological outcome of infection between WT and vhs mutants, at this point the exact mechanism (s) involved are not known. Since mutation of the vhs protein does not alter the tissue specificity or the neuroinvasiveness of BV, the vhs protein appears to be involved in the general replicative efficacy of BV and is not responsible for the extreme neurovirulence of BV.
-
The authors gratefully acknowledge the provision of anti-HSV vhs serum by Dr. D. Leib. This project was supported in part by PHS grants 2P40 OD010988 and 1P40 OD010431.
-
None of the authors have any competing interests. This article does not contain any studies with human subjects performed by the any of the authors.
-
D Black and R Eberle performed all experiments, JW Ritchey carried out pathological studies, and ME Payton performed all statistical analyses. All authors read and approved the manuscript.

 DownLoad:
DownLoad: