









HTML
-
In humans and other mammals successful defense against virus infection and protection from viral pathogenesis requires a careful coordination of innate and adaptive immunity. Innate immunity forms the first line of protection against invading viruses by utilizing germline encoded pattern recognition receptors(PRRs)to sense conserved pathogen associated molecular patterns(PAMPs)that are physiologic hallmarks of infection. Adaptive immunity is initiated soon after innate immunity is induced, and is programmed and modulated by the cells and products from the innate immune response. A surprising degree of complexity in crosstalk between the individual components of the innate immune system itself has emerged as an underlying factor in programming antiviral immunity. In particular, RNA viruses represent a wide range of pathogenic viruses, and they account for the majority of emerging and re-emerging viruses of public health importance. RNA viruses cause disease ranging from local inflammatory responses to hemorrhagic fever to cancer. This review presents general concepts of PRR function in viral infection, and then focuses on the molecular biology and virus-host interactions of RIG-I-like receptors(RLRs). RLRs are the major PRRs of RNA virus infection and also play a role in sensing RNA products of DNA virus infection. Special focus is given to the emerging complexity of crosstalk between the RLRs and other innate immune pathways and how regulation of the RLR pathway affects a diversity of outcomes.
-
Studies of RNA viruses have revealed mechanisms of immune programming, beginning with activation of the innate immune response. Initiation of the global immune response to virus infection begins with binding of a host PRR with its cognate viral PAMP in a reaction that is designed to distinguish self from nonself macromolecules to tell the body that it is indeed infected with a virus. Many distinct families of PRRs have been shown to play a role in detecting viral infection including toll-like receptors(TLRs), nucleotide-binding, oligomerization domain(NOD)-like receptors(NLRs), AIM2-like receptors(ALRs) and retinoic acid-inducible gene I(RIG-I)-like receptors(RLRs)(Takeda and Akira, 2004; Takeuchi and Akira, 2008; Elinav et al., 2011; Brunette et al., 2012). In addition, several other PRRs have also been shown to respond to nucleic acid products of viral infection such as protein kinase R(PKR), 2′-5′-oligoadenylate synthetase(OAS), cGAMP synthase(cGAS), non RLR-helicases, and class A scavenger receptors(SR-As)(Sen, 2000; Williams, 2001; DeWitte-Orr et al., 2010; Fullam and Schroder, 2013; Sun et al., 2013). A common principle of PRR function is that each PRR has the ability to discriminate between self vs. non-self during a viral infection despite the fact that viruses are obligate intracellular parasites and derived entirely from host cell components. This discrimination occurs due to either a unique identity of the viral PAMP or its aberrant location within a cell or animal. With a few notable exceptions, the viral PAMP is comprised of products from the viral nucleic acid genome, be they DNA products from a DNA virus infection or viral RNA from either an RNA virus or DNA virus expression products(Barbalat et al., 2009; Ichinohe et al., 2010). Following detection of a viral PAMP, PRRs typically initiate a signaling cascade that results in downstream transcriptional activation and gene expression of a variety of antiviral effectors, cell metabolism modulators, pro-inflammatory cytokines and chemokines, and initiation of adaptive immune programs.
In general, a fully functional or competent immune response will require the function of many different PRRs, each playing a temporal or tissue/cell-specific role in viral pathogen recognition and signaling of antiviral defenses for protection from a single pathogen. The requirement for pathogen sensing through multiple receptor pathways occurs despite the fact that these disparate sensors converge on many of the same signaling molecules and transcription factors. Experimental models of in vivo virus infection are largely dependent on the use of genetically inbred animals and optimized doses of infectious agent that lead to dramatic endpoints; for example mortality, specific pathology, or other directly observed outcomes. The reality though is that most natural human infection outcomes are less dependent on any one component, but compromise of a single aspect of human immunity may help explain the large spectrum of morbidity and mortality observed in clinical outcomes for a single virus infection.
-
The RIG-I-like receptors(RLRs)include three cytosolic PRRs that recognize RNA as PAMPs during viral infection; RIG-I, melanoma differentiation-associated factor 5(MDA5) and laboratory of genetics and physiology 2(LGP2)(reviewed in(Takeuchi and Akira, 2008; Loo and Gale, 2011))(Figure 1). Each is widely expressed by most cells and highly inducible in both a cell extrinsic(by type I IFN signaling) and/or cell intrinsic manner(direct virus-induced gene expression mediated by interferon regulatory factor(IRF)3 transcription factor signaling)(Yount et al., 2007).
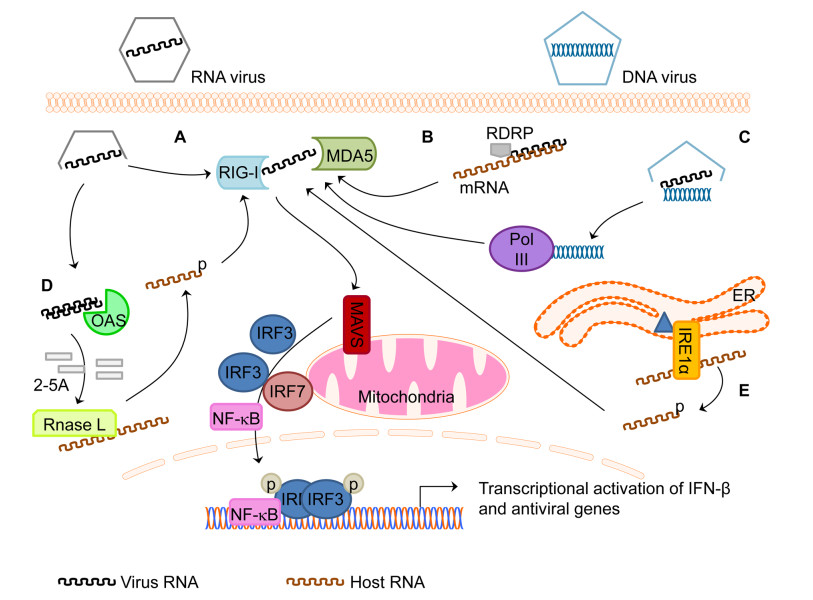
Figure 1. The RLR pathway is responsive to diverse stimuli to drive innate immunity.(A)Prototypic RLR pathway signaling is initiated following RIG-I or MDA5 binding to PAMP RNA from an RNA virus, undergoing a conformational change, interacting with MAVS, and activating a signaling cascade resulting in activation of transcription factors IRF3/7 and NF-κB.(B)Virus RDRP can use host RNAs as a template to generate 5′PPP dsRNA that serve as RLR PAMPs.(C)Cytoplasmic DNA from DNA viruses or bacteria is sensed by RNA Pol III and transcribed into RLR PAMP RNA.(D)OAS proteins sense dsRNA to generate 2-5A oligoadenylates leading to activation of RNase L and cleavage of RNAs resulting in RLR PAMP RNA bearing 3′ cyclic monophosphate.(E)Infection with bacteria can lead to the presence of bacterial toxins within the ER that activate the unfolded protein response(UPR)resulting in endonuclease IRE1α processing of host mRNA and generation of RLR PAMP RNA bearing 3′ cyclic monophosphate.
The three RLRs are ATPase dependent DExD/H RNA helicases and share homology throughout their central helicase domains as well as the C-terminal domain that has been described as a repressor domain(RD)(Saito et al., 2007; Cui et al., 2008; Takahasi et al., 2008). RIG-I and MDA5 possess two N-terminal caspase activation and recruitment domains(CARDs) and have been shown to recognize cytosolic RNA during infection to activate innate immune programs. LGP2 lacks the N-terminal CARD domains and is thought to function as a regulator of RIG-I and MDA5 signaling, although it may play other roles in regulating immune responses(discussed in further detail below)during virus infection(Saito et al., 2007; Venkataraman et al., 2007; Suthar et al., 2012).
RIG-I and MDA5 function as PRRs by surveillance of the intracellular environment for RNA PAMPs and undergo a conformational change upon binding RNA to expose their N-terminal CARD domains. The exposed CARD domains facilitate a homotypic protein: protein interaction with the CARD of the mitochondria associated membrane-bound, common adaptor MAVS(also known as IPS-1, CARDIF and VISA)(Meylan et al., 2005; Seth et al., 2005; Xu et al., 2005). The assembly of the RLR/MAVS complex recruits further members of the signal transduction cascade, including the protein kinases, TBK1 and IKKε, that direct the phosphorylation and activation of IRF3/IRF7, and the NF-κB kinase complex that activates and drives the nuclear translocation of NFκB and IRF3/IRF7. These processes result in the induction of pro-inflammatory genes, antiviral genes, and type I interferon(IFN). Secreted IFN then signals through its specific receptor and down through the JAK-STAT pathway to induce the expression of hundreds of interferon stimulated genes(ISGs). ISGs have antiviral and other properties that serve to restrict virus replication. A combined model of RIG-I activation proposes that RIG-I actively surveys RNAs as an ATPase-dependent translocase(Myong et al., 2009). Following encounter with specific PAMP ligand properties, RIG-I undergoes a conformational change and associates into homo-oligomers that are brought to the intracellular surface of mitochondria, mitochondrial-associated membranes(MAMs) and peroxisomes by molecular chaperones such as TRIM25 and 14-3-3ε(Saito et al., 2007; Dixit et al., 2010; Horner et al., 2011; Liu et al., 2012). Less is known about the specific events regulating MDA5 activation following RNA binding, but accumulating evidence suggests the signaling competent form of MDA5 is a large oligomer of multiple MDA5 molecules arranged along ligand PAMP RNA so that the CARD domains are exposed for interaction with MAVS(Peisley et al., 2011; Berke and Modis, 2012). Many distinct cellular factors and processes have been shown to regulate this signaling cascade and are recently reviewed elsewhere(Wu and Chen, 2014).
-
RIG-I and MDA5 have been reported to detect both distinct and overlapping groups of viruses(Kato et al., 2006; Mibayashi et al., 2007; McCartney S. A. et al., 2008; Pichlmair et al., 2009). Briefly, RIG-I has been shown to be required for response to viruses including Paramyxoviridae family members, influenza and hepatitis C virus(HCV). MDA5 is essential for response to Picornaviridae family members such as EMCV. West Nile virus(WNV), Dengue virus, reovirus and LCMV have been reported to be detected by both MDA5 and RIG-I(Kato et al., 2006; Loo et al., 2008; Zhou et al., 2010). However, subsequent work has shown that Sendai virus(originally thought to be solely sensed by RIG-I)specifically antagonizes MDA5 signaling, while EMCV(originally thought to be a MDA5 dependent virus)targets RIG-I for degradation, raising the possibility that both RLRs may be able to detect a wider array of viruses than initially appreciated(Andrejeva et al., 2004; Barral et al., 2009). The importance of the RLRs in immunity against viruses is highlighted by the multiple ways these viruses have evolved to directly antagonize RLR signaling. In addition to the above examples, the NS1 protein of influenza binds RIG-I and the V proteins of Paramyxoviridae family members efficiently bind to both MDA5 and LGP2 to prevent RLR signaling(Mibayashi et al., 2007; Parisien et al., 2009). The HCV NS3/4A protein disrupts RLR signaling by proteolytically cleaving MAVS from the surface of intracellular membranes(Meylan et al., 2005; Loo et al., 2006).
Studies that characterize PAMP ligands of RIG-I have demonstrated that non-self recognition depends on several properties of viral RNA including PAMP motif length, structure, modification, and composition(Hornung et al., 2006; Marques et al., 2006; Saito et al., 2008; Schlee et al., 2009). The most consistent observation from these studies is that RIG-I ligand RNA requires a 5′ppp in conjunction with at least a small amount of secondary dsRNA structure. Importantly though, some RNAs that contain both 5′ppp and dsRNA do not stimulate RIG-I-dependent signaling, suggesting that sequence composition is another incompletely understood determinant of RIG-I PAMP specificity(Saito et al., 2008; Schnell et al., 2012). The 5′ppp physically interacts with the C-terminal RD of RIG-I while the helicase domain makes contact with dsRNA(Cui et al., 2008; Jiang et al., 2011).
The precise nature of the viral ligand for MDA5 has not been well described. MDA5 ligand specificity has been reported be either long dsRNA(> 2 kilobases(kb)), or that ligand RNA not only requires long dsRNA but "higher order" molecular complexes of RNA containing both dsRNA and ssRNA(Kato et al., 2008; Pichlmair et al., 2009). The question of how MDA5 discriminates between long dsRNAs, short dsRNAs, and other higher order complexes is important because it is likely that there are several unidentified factors that define the viral ligand of MDA5 just as there are for RIG-I. Indeed, two recent reports suggest that MDA5 is capable of signaling in response to RNA agonists as short as ~ 200 nucleotides(Deddouche et al., 2014; Runge et al., 2014). Deddouche et al. demonstrate that a short, MDA5 stimulatory RNA is actually bound by LGP2 during EMCV infection which then enables MDA5 mediated signaling through incompletely understood mechanisms resulting from the LGP2/MDA5 interaction(Deddouche et al., 2014). Runge et al. demonstrate that short AU rich sequences derived from measles virus are capable of binding to both RIG-I and MDA5 and stimulating MDA5-dependent innate immunity in a phosphate independent manner(Runge et al., 2014). Another PAMP determinant not directly related to length or secondary structure is that the lack of 2′0 methylation on coronavirus RNA was shown to be necessary to induce MDA5 dependent innate immune signaling(Zust et al., 2011).
Nikonov et al. showed that a PAMP of non-viral genomic origin was detected by both RIG-I and MDA5(Nikonov et al., 2013). These authors used extensive biochemical experiments to demonstrate that the Semliki Forest virus RNA dependent RNA polymerase(RDRP)can use host RNA as a template to generate 5′ppp containing dsRNA products that serve as stimulators of both RIG-I and MDA5. Future work to sequence RLR associated RNAs and expand these findings to other virus infection models will inform our understanding of the biological significance of this novel host-derived PAMP. It is worthwhile to note that when expressed alone in human cells, RNA-dependent RNA polymerases from other positive sense RNA viruses have been shown to induce IFN, suggesting that generating RNA PAMPs from viral polymerase products of host genetic material could be a more general phenomenon that the host cell has leveraged to program the innate immune response to virus infection(Moriyama et al., 2007).
-
RIG-I and MDA5 have been shown to be the two major PRRs responsible for sensing cytoplasmic viral RNA and activating innate immunity in a MAVS-dependent manner(Figure 1). Of note is that extracellular administration of RNA has been shown to stimulate an RLR dependent response, and that intracellular viral RNA ligand s are frequently sequestered in a membranous web. Moreover, MDA5 itself has been described to have a low affinity for ligand RNAs(Hoenen et al., 2007; Longhi et al., 2009; Takahasi et al., 2009). These observations suggest that additional RLR cofactors of PAMP sensing may influence RLR function.
The OAS family of proteins are cytosolic PRRs that bind to dsRNA. This binding of dsRNA activates the OAS protein to catalyze the production of oligoadenylates with an atypical 2′, 5′ linkage known as 2-5A. The RNase L protein, a nonspecific RNA nuclease becomes activated by binding to 2-5A products upon which it cleaves viral and host RNAs. The RNase L cleaved RNAs contain a 2′, 3′-cyclic phosphate at the 3′ end that has been shown to be a RLR PAMP(Malathi et al., 2007; 2010). OAS thus functions as a PRR cofactor within the RLR pathway to amplify innate immune signaling. Future work will continue to reveal the significance of RNase L cleaved RNAs in antiviral immunity and if there are other host RNA modification pathways that can lead to the amplification of RLR mediated innate immune signaling.
HMGB protein family members were long known to be nuclear, DNA binding structural proteins, but HMGB1 was also discovered to be actively secreted by cells during acute virus infection(Andersson et al., 2002). Yanai et al. then further defined a role for the HMGB family members as "universal sentinels" of both DNA and RNA PAMPs in promoting TLR and RLR signaling(Yanai et al., 2009). HMGB proteins may therefore be responsible for delivering nucleic acid from the extracellular space to endosomes and the cytoplasm. However, this simple model is complicated by the fact that HMGB expression also enhanced signaling to nucleic acid transfected directly into the cytoplasm. Additionally, SR-A proteins are cell surface receptors that appear to play a more clear role in facilitating dsRNA entry into the cytoplasm and endosomes for recognition by the RLRs and TLR3, respectively(DeWitte-Orr et al., 2010). It is interesting to note that while poly(I:C)efficiently induces signaling through both RIG-I and MDA5 when transfected directly into cells, MDA5 appears to play a dominant role in modulating the adjuvant effects of poly(I:C)when it is administered in vivo in the absence of any transfection reagent(Longhi et al., 2009; Takahasi et al., 2009; Wang et al., 2010). One hypothesis put forth to explain this observation is that accessory factors such as HMGB proteins or SR-As may preferentially bind and then present extracellular nucleic acid ligand s to the intracellular/ cytoplasmic environment in specific cell types for MDA5 mediated PAMP recognition and induction of the innate immune response.
Several non-RLR helicases have also been implicated in antiviral immunity with some appearing to play a role in regulating the RLR pathway(reviewed in(Fullam and Schroder, 2013)). DDX3 was identified as a binding partner of MAVS and facilitated a full IFN response following RNA stimulation of cells(Oshiumi et al., 2010). It is unclear what exact role DDX3 is playing in innate antiviral immunity though, since despite being an RNA binding protein and MAVS binding partner, this helicase was also identified as a host susceptibility factor during HCV and HIV infection(Yedavalli et al., 2004; Ariumi et al., 2007). DHX9 was similarly shown to bind dsRNA and be essential for full innate immune signaling through MAVS, but has also been implicated as a host susceptibility factor to virus infection(Zhang et al., 2011). DDX60 was identified as a more upstream cofactor of the RLR pathway to promote full innate immune signaling by binding RNA and physically associating with the RLRs but not with downstream signaling components(Miyashita et al., 2011). The antiviral stress granule has been proposed as an intracellular structure critical for facilitating RLR pathway activation by bringing together RNA ligands and signaling components(Onomoto et al., 2012). DHX36 promotes RLR signaling by binding to dsRNA and the protein kinase R(PKR)to form the antiviral stress granule that is thought to be a platform for RLR-mediated signaling in certain cell types(Yoo et al., 2014). Future work is needed to ascertain the function of these RNA binding proteins in the RLR pathway and their role as ligand chaperones, binding cofactors, or signaling enhancers.
-
The RLR pathway plays an immune regulatory role by indirectly interacting with other signaling pathways to alter the overall gene expression profile of cells. Interaction with inflammasome signaling pathways is one such example. The inflammasome is a multiprotein complex that includes a NOD-like receptor(NLR), the ASC adaptor protein, and caspase 1. This complex becomes activated in response to certain inflammatory stimuli and functions to induce the expression and secretion of interleukin 1(IL-1)family cytokines including IL-1β and IL-18 that drive a potent inflammatory response. Inflammasomes are named based on the nature of their initiator NOD-like protein(Schroder and Tschopp, 2010). RLR mediated signaling can increase inflammasome activity in a cell intrinsic manner and may also indirectly decrease inflammasome activity through IFN actions that repress the activity of NLRP1 and NRLP3, and by decreasing the abundance of substrate and pro-IL-1β(Guarda et al., 2011). The complex interaction of RLRs and inflammasomes is further underlined by the fact that co-treatment of cells with both IFN-β and IL-1β results in increased ISG expression and increased control of West Nile virus infection compared to IFN-β treatment alone(Ramos et al., 2012).
Components of the RLR pathway can also directly interact with factors in inflammasome and caspase signaling to alter cell intrinsic responses. Poeck et al. showed that RIG-I interacts with CARD9 and BCL10 in a MAVS dependent manner to activate NF-κB and induce pro-IL-1β expression(Poeck et al., 2010). Additionally, the same authors also showed that activated RIG-I could interact with the inflammasome adaptor protein ASC to directly induce caspase 1 dependent cleavage and release of mature IL-1β. RLR signaling through MAVS was shown to induce apoptosis in a caspase-9 and Apaf-1dependent manner(Besch et al., 2009). MAVS may also play a role in activating inflammasome and caspase signaling independent of signaling via RIG-I and MDA5. The chemical actinomycin D was shown to be capable of inducing apoptosis in a MAVS dependent mechansim in the absence of any apparent RLR ligand (Guan et al., 2013). Additionally, MAVS was required to recruit NLRP3 to mitochondrial membrane surfaces for efficient production of mature IL-1β in response to ATP, and this inflammasome activation was independent of type I IFN induction(Subramanian et al., 2013). Thus, RLR signaling can play an important pro-inflammatory role to control virus infection and support the antiviral immune response.
-
An unexpected role for the RLR pathway was initially discovered in 2007 and further elucidated in 2009 when it was demonstrated that the dsDNA poly(dA-dT) and several DNA viruses can induce an IFN response through the RLR pathway(Cheng et al., 2007; Ablasser et al., 2009; Chiu et al., 2009). This RLR sensing of DNA is accomplished when poly(dA-dT)or viral substrate DNA is transcribed by DNA-dependent RNA polymerase III(Pol III)into a RIG-I stimulatory, 5′ triphosphate containing RNA ligand (Figure 1).
The recent identification of the cGAS/cGAMP pathway has increased our understanding of the intracellular sensing of DNA as a PAMP and the activation of innate immune programs against DNA viruses(Sun et al., 2013; Wu et al., 2013). The current model of cGAS function in innate immunity proposes that cytosolic dsDNA is engaged by cGAS which undergoes ligand -induced oligomerization(Li et al., 2013). The catalytically active oligomer of cGAS then synthesizes the endogenous second messenger cGAMP. cGAMP binds the adaptor protein STING to induce signal transduction that leads to the nuclear translocation and activation of several transcription factors including IRF3(Ablasser et al., 2013; Gao et al., 2013; Zhang et al., 2013). Remarkably, several studies have now indicated that this pathway may contribute to intracellular RNA sensing and /or protection against RNA virus infection, but the mechanism remains unclear(You et al., 2013; Schoggins et al., 2014). In vitro transfection experiments seem to clearly demonstrate unique and non-overlapping roles for components involved in sensing DNA and RNA(Brunette et al., 2012). The respective roles of these two pathways in activating innate immunity can, at least partly, be explained by two separate phenomenon. First, in vitro over-expression of components from either the RNA or DNA sensing pathways can stimulate innate immune signaling in a ligand -independent manner. Furthermore, genetic ablation of the cGAS DNA sensing pathway appears to alter basal innate immune gene expression of cells(Schoggins et al., 2014). This observation is important in the consideration of host defense because tonic IFN priming of cells has been shown to be an important component of the innate immune response to virus infection, and IFN priming has been known for a very long time to inhibit both RNA and DNA virus infection(Isaacs et al., 1958; Hata et al., 2001). Future research to clearly establish the mechanism of crosstalk between DNA and RNA sensing pathways will provide exciting insight into immunity to virus infection.
-
Experimental infection of mice with West Nile virus(WNV), an emerging RNA virus, has revealed many key aspects of host-pathogen interactions in an animal model that closely recapitulates the human course of disease(reviewed in(Suthar et al., 2013)). Several studies have clearly established an important role for MAVS, the central signaling adaptor of the RLR pathway, in defense against WNV infection(Fredericksen et al., 2008; Suthar et al., 2010; Suthar et al., 2012). Recent work extended our understanding of the individual and combined roles of RIG-I and MDA5 in pathogen recognition and immunity to WNV infection using WT and RLR knockout(KO)mice and cells. Each RLR individually contributes to pathogen recognition and immune protection against WNV in vivo and in vitro. RIG-I and MDA5 were shown to detect distinct PAMPs with differential kinetics during the course of WNV replication to mediate complementary, nonredundant roles in viral detection and innate immune gene induction(Errett et al., 2013).
Loss of both RLRs during in vivo challenge of mice with WNV reveals a susceptibility phenotype identical to animals lacking MAVS(Suthar et al., 2010). The identical magnitude and kinetics of mortality in these double KO mice occurred despite expression of all other related and unrelated PRRs. In vitro infection of key target cells of infection further revealed that innate immune signaling in RIG-I and MDA5 double KO cells was identical to MAVS KO cells. Therefore, while other PRRs may function as accessory partners in RLR signaling, RIG-I and MDA5 are the two essential PRRs responsible for transducing innate immune signaling in this model. Efficient and early PAMP recognition is required to induce an IRF3-dependent gene expression signature that controls viral replication in a cell-intrinsic manner(Daffis et al., 2007). In cells lacking RIG-I expression, WNV replication proceeds at a higher rate despite the ability of these cells to mount a robust innate immune response at late times of infection. MDA5 deficiency in MEFS reveals a lack of IRF7-driven IFN-α2a expression at later time points without an increase in viral replication. Lack of innate immune diversification and amplification in the MDA5 KO MEFs cells without a concommitant increase in virus may be due to the ability of pathogenic WNV to block type I IFN signaling when MDA5 PAMPs accumulate (Keller et al., 2006; Daffis et al., 2008; Loo et al., 2008; Laurent-Rolle et al., 2010).
In addition to inducing effector genes that are antiviral on a cell-intrinsic level, the central role of the innate immune response in programming an effective adaptive response in the context of WNV is highlighted by several key studies. As previously noted, MAVS deficiency in vivo results in lethality and increased viral replication, but also a dysregulated adaptive response. Despite an increase in most inflammatory pathways and immune cellularity, anti-WNV antibodies had reduced function and regulatory T cells were reduced in number(Suthar et al., 2010). Interestingly, deficiency in the RLR family member LGP2 did not greatly affect observed innate immune gene induction, but instead lead to an increased lethality in mice that was attributed to a CD8 T cell-intrinsic defect(Suthar et al., 2012). The lack of MDA5 alone in vivo lead to a deficient CD8 T cell response in the CNS, but this was not a cell-intrinsic phenotype(Lazear et al., 2013). Loss of inflammasome signaling also lead to an increased lethality in mice that was linked to a lack of CD8 T cell function in the CNS(Ramos et al., 2012). Innate immune signaling thus regulates both cell intrinsic immune responses as well as adaptive immunity to program successful defense against WNV.
It is important also to note a role for RLR signaling for immune response integrity during conditions of polymicrobial infection in which a host is coinfected with virus and bacteria. Such coinfection can lead to stimulation of multiple innate immune pathways and unexpected modulation of disease outcomes. Infection with vesicular stomatitis virus(VSV)was shown to induce a RLR-dependent IRF3 activation that suppressed the development of TH1 and TH17 T cells in the context of a subsequent bacterial infection(Negishi et al., 2012). This RLR suppression of specific T cell development was due to cross-interference of TLR signaling and mice that were infected with VSV were found to be mortally susceptible to an otherwise sub-lethal dose of bacteria.
We also briefly note that mutations within RIG-I and MDA5 have been strongly associated with multiple autoimmune disorders in humans through genome wide association screens(reviewed in(Loo and Gale, 2011)). How these mutations influence disease outcomes in humans has not been fully elucidated. In vitro assays demonstrate that autoimmune-associated RLR variants exhibit both loss and gain of function(Shigemoto et al., 2009; Funabiki et al., 2014). Future work will continue to characterize the functional signaling consequence of autoimmune-associated RLR variants as well as the contribution of endogenous or microbial ligands to the development of disease.
-
Hepatitis C virus(HCV)is an RNA virus and member of the hepacivirus family. It is a cousin of WNV, being part of the same viral genera-the Flaviviridae. However, HCV is a blood-borne human virus whereas WNV is transmitted by the bite of an infected mosquito. Other differences between these two viruses include that WNV is neurotropic whereas HCV is hepatotropic to infect hepatocytes within the liver, and that WNV typically mediates an acute/self-limiting infection whereas HCV most often mediates a chronic infection. Notably, chronic HCV infection is a leading etiology of liver cancer called hepatocellular carcinoma(Horner and Gale, 2013). HCV infection is recognized by RIG-I to drive a hepatic innate immune response capable of suppressing infection such that 25%–30% of acute HCV infections spontaneously resolve. HCV is recognized by RIG-I through a combination of 5′ triphosphate and a poly-uridine/cytosine(poly-U/UC)motif present within the 3′ nontranslated region of the viral RNA(Saito et al., 2008; Schnell et al., 2012). In vitro studies to model acute HCV infection revealed that HCV is sensed by RIG-I early but that the resulting innate immune response is shut down within 24–48 hours after infection, thus allowing the virus to replicate and spread unimpeded by the innate immune response of the host cell(Meylan et al., 2005; Loo et al., 2006). It was discovered that the RLR pathway was shut down by the actions of the viral nonstructural(NS)3/4A protease that targets MAVS on the mitochondria associated membrane to thereby ablate the RLR pathway signaling(Meylan et al., 2005; Loo et al., 2006; Horner et al., 2011). An important observation is HCV replicons with mutations in NS3 or NS4A, such that the virus cannot target or cleave MAVS, are effectively suppressed by the antiviral actions of the RIG-I pathway and are thus cleared from the cell(Horner et al., 2012). These observations indicate that viral outgrowth toward establishment of chronic HCV infection is highly dependent on HCV NS3/4A targeting and cleaving MAVS to inactivate the RLR pathway during acute infection. Overall, this outcome-dependent regulation of RIG-I signaling of innate immunity extends to provide a foundation for HCV persistence that is ultimately associated with the development of liver cancer. To this end the lack of control of HCV infection through viral evasion of RIG-I signaling and innate immunity are implicated indirectly in the development of HCV-related liver cancer.
HCV replicates as a population of related but genetically distinct viral quasispecies in which viral genetic diversity is attributed to the error-prone replication that is typical of RNA viruses, as the RDRP does not have proof reading function. In this respect the quasispecies complexity of the transmission-founder virus of the initial acute infection is a critical determinant of infection outcome, as the best-adapted viral genome will then replicate, further adapt, and persist in the new host. Genetic diversity within the NS3/4A protease coding region as well as in the poly-U/UC PAMP motif might therefore impact viral regulation of the RIG-I pathway to determine viral outgrowth through NS3/A targeting of RIG-I or viral clearance through effective poly-U/UC PAMP binding and signaling of a robust innate immune response. Recent work has revealed that striking genetic diversity exists among transmission/founder virus poly-U/UC motif such that differential recognition of the acute infection by RIG-I might be implicated in differential outcomes of infection(Stoddard et al., 2015).
-
In addition to their role in autoimmunity and the indirect regulation of virus-associated outcome to cancer, the RLRs have now been implicated directly or indirectly in the detection and/or disease outcome in response to RNA virus, DNA virus, bacterial and fungal infections(Figure 1). Given this central role of the RLRs, they can be considered as therapeutic targets to treat infections or pathologies for which there is no effective treatment. The ability to selectively activate or antagonize the appropriate PRR pathway as a directed therapy is attractive. One of the major targets of RLR activation, type IFN, has been used as an effective antiviral agent for decades (Lange et al., 2013). However, therapy with other pharmaceutical agents that induce interferon have been shown to be effective when type I IFN fails(Ferenci et al., 2008). This suggests that stimulation of multiple pathways, including type IFN receptor signaling, can be an effective strategy. RLR stimulation has been shown to lead to type IFN production and signaling, NF-κB activation, and inflammasome activation. Such pleiotropic responses could be highly beneficial when a more limited therapy fails. Thus, RLR-targeted therapeutic strategies could include use as direct antivirals or as new adjuvants for vaccines. The innate/adaptive immune interface has been highlighted as key to effective immunity(Iwasaki and Medzhitov, 2010), and RLR signaling and functional interface with the adaptive immune response underscores the attractiveness of targeting RLRs for immune enhancing and antiviral therapies(Longhi et al., 2009; McCartney et al., 2009; Suthar et al., 2010; Suthar et al., 2012; Lazear et al., 2013). Approved adjuvants such as alum and MPL may not accurately represent the appropriate innate immune stimulation for a natural course of infection for the pathogen being immunized against. Combinatorial adjuvants activating the appropriate innate immune pathways may be critical to successful vaccine design(Kasturi et al., 2011) and should therefore consider RLR agonists as possible adjuvants. Continuing research into the emerging complexity of the initiation and functional consequences of the RLR pathway will enable us to design these rational therapeutic strategies.
-
MG and JE are supported by National Institutes of Health grants AI083019, AI104002, and AI88778.
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: