










HTML
-
Japanese encephalitis (JE) is a severe viral encephalitides that is prevalent in Eastern and Southern Asia and the Pacific Rim, with a reported 50, 000 cases and 15, 000 deaths annually (Misra and Kalita, 2010). Due to recent changes in the distribution and epidemiology of the virus, JE has become a growing public health concern worldwide (Solomon and Winter, 2004; Ghosh and Basu, 2009).
The pathogen of JE is termed JE virus (JEV), which belongs to the genus Flavivirus in the family Flaviviridae. The JEV genome is composed of single-stranded RNA of ~11 kb in length, comprising a 5′-untranslated region (UTR), a single open reading frame (ORF), and a 3′-UTR (Chambers et al., 1990). The single ORF encodes three structural proteins, including capsid protein (C), precursor membrane protein (prM), and envelope protein (E), and seven nonstructural (NS) proteins, including NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. The E protein is the major antigenic determinant, and is also involved in viral attachment, fusion, penetration, hemagglutination, host range and cell tropism, and virus virulence and attenuation (Wu et al., 2003). According to its crystal structure, the ectodomain of E protein is composed of three discernible domains. Domain Ⅰ (DI) folds into a nine-stranded β-barrel and is located in the center of the protein. Domain Ⅱ (DII) is dimerized by two extended loops protruding from DI, with a conserved fusion peptide at its tip. Domain Ⅲ (DIII) has an immunoglobulin-like β-barrel structure, and is connected to DI by a flexible peptide linker (Luca et al., 2012). A flexible helical stem is found close to the C-terminus of DIII, which plays an essential role in the zippering reaction during membrane fusion (Pangerl et al., 2011). The C-terminal transmembrane helices then serve to anchor E protein to the viral membrane (Mukhopadhyay et al., 2005). Twelve strictly conserved cysteines within E protein were identified to form six intramolecular disulfides, which are essential for stabilizing the tertiary structure of the protein (Luca et al., 2012).
The last few decades have seen great progress in the development of reverse genetic approaches for studying various aspects of virus biology and pathogenesis, as well as for designing new vaccines (Yun and Lee, 2014). However, the toxicity of full-length cDNA in bacteria has greatly hindered the generation of JEV infectious clones (Ward and Davidson, 2008). Various strategies have been employed to overcome this problem, including the use of very-low-copy-number vectors (Sumiyoshi et al., 1995; Zhang et al., 2001), the use of specific Escherichia coli strains or yeast as hosts (Sumiyoshi et al., 1995; Polo et al., 1997), insertion of short introns into unstable regions of virus cDNA (Yamshchikov et al., 2001), suppression of cryptic bacterial promoters in the flaviviral genome (Pu et al., 2011; Pu et al., 2014), split-up of the genome as bicistronic constructions (Orlinger et al., 2006), and separation of the genome into two parts followed by in vitro ligation and RNA transcription (Sumiyoshi et al., 1992). More recently, an infectious subgenomic amplicons method was proposed, in which transfection of overlapping double-stranded DNA fragments covering the entire genome of an RNA virus into susceptible animal cells is used to synthesize a DNA copy of the complete viral genome by in cellulo recombination (Aubry et al., 2014).
In vitro ligation methods for the generation of flaviviral infectious cDNA are continuously being developed. For example, the genome of West Nile virus (WNV) lacking structural genes under the control of a cytomegalovirus (CMV) promoter was ligated in vitro to complementary DNA fragments harboring wild type or mutated structural genes, and then the ligation mixture was directly transfected into permissive cells to recover wild type or mutated infectious viruses (Lin et al., 2012). Although this method could successfully achieve the rapid generation of flaviviral infectious clones, it also introduced synonymous mutations to generate unique restriction endonuclease sites, which could lead to unpredictable consequences, especially if the mutated nucleotide plays an essential role in RNA replication or gene expression (Yun et al., 2009; Melian et al., 2010).
Herein, we describe a modification of this in vitro ligation method and successfully established a reverse genetic system for the rapid mutagenesis of the structural genes of JEV. Our method is based on an In-Fusion cloning (in vitro recombination) system instead of traditional endonuclease-dependent ligation, in order to avoid the generation of any unnecessary mutation and to allow for the generation of viruses that accurately represent native or site-mutated viral sequences. Using this approach, we determined the key role of the conserved cysteine residues of the JEV E protein in the virus life cycle.
-
Baby hamster kidney cells BHK-21 (preserved in "Chinese general virus collection center" (CGVCC)) were cultivated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in 5% CO2 at 37 ℃. The JEV strain SA14 was recovered from pACYC-JEV (kindly donated by Prof. Bo Zhang, Wuhan Institute of Virology, China) as previously described and stored at -80 ℃ (Li et al., 2014). JEV-specific antibodies and anti-EDIII and anti-NS1 were used as previously described (Du et al., 2015).
-
The full-length JEV infectious cDNA clone pACYC-JEV was modified (Li et al., 2014). Firstly, the 5′-terminal nucleotides (1–172 bp) of the virus genome were introduced immediately downstream of the CMV promoter of pEGFP-C1 (Clontech, CA, USA) by overlap-extension polymerase chain reaction (OE-PCR) to generate pJEV-5UTR, and the nucleotides corresponding to the enhanced green fluorescent protein (egfp) gene was removed. Then, the nucleotides of the virus genome from nucleotide 2560 to the 3′-terminus as well as nucleotides encoding the ribozyme of hepatitis delta virus (HDVr) and the polyadenylation (poly A) sequence of Simian virus 40 (SV40) at the 3′-terminus of the genome were inserted downstream of the 5′-terminal nucleotides of pJEV-5UTR, generating the pJEV-infu backbone, which lacks nucleotides 173–2559 corresponding to the entire sequences of the prM and E proteins, as well as small portions of the C and NS1 proteins. Note that the engineered site where nucleotide 172 was ligated to nucleotide 2560 directly generates a new unique endonuclease Sma I site, which could be used for the linearization of pJEV-infu.
To generate mutated viruses using the pACYC-JEV infectious clone system, mutations were introduced into the pACYC-JEV plasmid by OE-PCR as described previously (Li et al., 2014).
-
The JEV-complement fragment was generated using pACYC-JEV as a template and the primers infusion-F and infusion-R (Table 1). The resulting DNA fragment encodes nucleotides 158 to 2574 of JEV SA14, corresponding to the entire missing sequence region in pJEV-infu as well as 15 nucleotides extending at the 5′-terminus and 3′-terminus, respectively.

Table 1. Primer sequences used in this study.
OE-PCR was performed to introduce mutations into the JEV-complement fragment (Lin et al., 2012). Briefly, each desired mutation was introduced into two partially overlapping "inner primers." A left-PCR was performed using infusion-F as a common forward "outer" primer and the antisense inner primer, whereas a right-PCR was performed including a sense inner primer and a common reverse "outer" primer, infusion-R. After gel purification, these two first-round PCR products were used together as a template for a second PCR, employing the forward and reverse "outer" primers described above.
-
The backbone plasmid pJEV-infu was linearized by Sma I digestion, and the JEV-complements were ampli fied by PCR. The products were then gel-purified and in vitro recombination reactions were performed by incubating the linearized backbone (0.2 μg) and the complementary fragment (0.2 μg) with 2 μL of 5× In-Fusion HD Enzyme Premix (Clontech, CA, USA) in a final volume of 10 μL according to the manufacturer's instructions. The entire recombination reaction mixture was directly transfected into BHK-21 cells (5 × 105 cells/well) in a six-well plate using Lipofectamine 2000 (Invitrogen, CA, USA) according to the manufacturer's instructions. Transfected cells were cultured, and virus-containing supernatants were harvested at 2 to 5 days post transfection (p.t.). After centrifugation at 500 × g for 10 min to remove cell debris, the supernatants were frozen at -80 ℃ until use.
-
BHK-21 cells (1 × 105 cells/well) grown on 24-well plates were infected with the supernatants of transfected cells obtained at 5 days p.t. as described above. At 36 h post infection (p.i.), the cells were fixed with paraformaldehyde (4% in phosphate-buffered saline [PBS]) for 15 min and then treated with PBS containing 0.5% Triton X-100 for a further 15 min at room temperature. Permeabilized cells were then stained with a JEV NS1-specific primary antibody (anti-NS1) and goat-anti-rabbit (fluorescein isothiocyanate [FITC]-conjugated; Proteintech, Chicago, USA) immunoglobulin.
-
BHK-21 cells (2 × 105 cells/well) grown on 12-well plates were infected with the supernatants of the transfected cells obtained as described above. At 36 h p.i., the cells were lysed and prepared for sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by transfer to a poly (vinylidenefluoride) membrane using a Trans-Blot apparatus (Bio-Rad, CA, USA). Western blot was performed with JEV-specific antibodies (anti-EDIII and anti-NS1) as primary antibodies and horseradish peroxidase-conjugated goat anti-rabbit antibody (Proteintech, Chicago, USA) as a secondary antibody. The antibody–protein complexes were visualized with SuperSignal West Pico Chemiluminescent Substrate (Fermentas, CA, USA) and the MicroChemi Bio-imaging System (DNR, IN, USA).
-
Monolayer cultures of BHK-21 cells (2 × 105 cells/ well) grown in 12-well plates were inoculated with sequentially diluted JEV. After 2 h of incubation at 37 ℃, the cells were washed with 1 mL DMEM three times and then coated with a layer of 1% SeaPlaque agarose (Lonza, Switzerland) in 2% FBS-DMEM. At 3 days p.i, 0.1% crystal violet solution was added to visualize the plaques.
-
Virus titers were determined based on the 50% tissue culture infectious dose (TCID50) using standard cell culture procedures. Briefly, BHK-21 cells were grown in 96-well plates, and six replicates were infected with 100 μL of 10-fold serial dilutions of the virus sample. After incubation at 37 ℃ for 3 days, the cytopathic effect (CPE) was monitored using an inverted microscope, and infectivity titers were expressed as TCID50/mL according to the method previously described (Reed and Muench, 1938).
-
BHK-21 cells were infected with the indicated viruses at a multiplicity of infection (MOI) of 0.01. Cell-free media were collected at 12, 24, 36, 48, and 60 h p.i., and virus titers were determined as described above. Each virus infection was performed in triplicate.
-
BHK-21 cells transfected with ligation reaction mixtures were harvested at 5 days p.t., and total RNA was extracted using TRIzol reagent (Invitrogen, CA, USA). One-step qRT-PCR was then carried out using One Step SYBR PrimeScriptTM PLUS RT-PCR Kit (Takara, Japan) following the manufacturer's instructions. Primers targeting the NS1 regions of JEV were used, including the forward primer JEV qRT-F and the reverse primer JEV qRT-R (Table 1). The amount of JEV genome was normalized to the β-actin gene using the comparative cycle threshold values determined in parallel. Primer sets targeting β-actin included the forward primer actin-F and reverse primer actin-R (Table 1). PCR specificity was verified by melting curve analysis.
-
In vitro RNA transcription was performed as previously described (Li et al., 2014). Briefly, the wild-type or mutated infectious clone pACYC-JEV plasmids were digested with Xho I and purified, and were then subjected to in vitro transcription using a MEGAscript T7 kit (Ambion, TX, USA) according to the manufacturer's protocols. The RNA was resolved in RNase-free water and stored at -80 ℃. BHK-21 cells (1 × 105 cells/well) growing in a 24-well plate were transfected with 200 ng wild type or mutated transcripted viral RNA. At 48 h p.t., the cells were fixed and stained with anti-NS1 antibody.
Cells, viruses, and antibodies
Plasmid construction
Generation of the wild type or mutated JEV-complement
Virus production
Immunofluorescence assay (IFA)
Western blot analysis
Plaque-forming assay
Viral titer determination
Viral growth curve analysis
Quantitative reverse-transcription (qRT)-PCR
In vitro RNA transcription and transfection assay
-
The major unstable region of JEV cDNA in bacte-ria was previously identified to localize at the end of E protein and the beginning of NS1 (Yamshchikov et al., 2001). As shown in Figure 1A, a truncated form of the JEV genome lacking nearly all of the structural genes and a portion of NS1 (nucleotides 173 to 2559) was placed immediately downstream of the CMV promoter to construct a single-backbone plasmid (pJEV-infu), which could be stable in bacteria. Ligation of nucleotides 172 and 2560 generated a novel endonuclease site, Sma I, which could be used to linearize the backbone plasmid to facilitate subsequent procedures. The HDVr and the poly A signal from SV40 adjacent to the 3′-end of the genome could ensure the correct processing of the 3′-terminus.
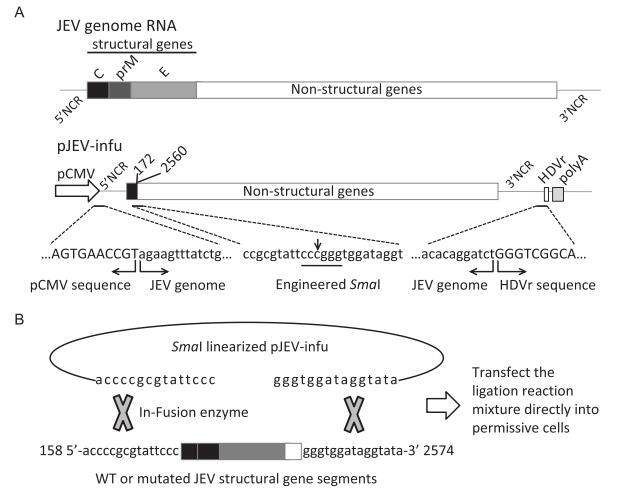
Figure 1. In vitro recombination-based strategy for the production of infectious JEV. (A) Schematics of the pJEVinfu backbone construct. A truncated form of the virus genome lacking structural genes (nucleotides 173 to 2559) was constructed under the control of CMV promoter. The HDVr and SV40 poly A were placed downstream of the viral 3′ UTR to ensure correct processing of the 3′-terminus of the genome. Note that the engineered endonuclease site Sma I was generated from ligation of nucleotide 172 with 2560. The sequences of CMV promoter and HDVr, together with the viral sequences were also indicated. (B) The entire complementary sequence carrying wild type or mutated structural genes was extended with 15 nucleotides at the 5′-terminus and 3′-terminus, respectively (JEV-complement), which overlapped with Sma I-linearized pJEV-infu at the ends, thus ensuring in vitro recombination using the In-Fusion cloning system. The recombination reaction mixture was then directly transfected into permissive cells to generate infectious virus.
The entire deleted portion with 15 nucleotide extensions at both the 5′-terminus and 3′-terminus was amplified by PCR, where mutations could be introduced easily using OE-PCR, and was then cloned into the linearized backbone plasmid pJEV-infu with the In-Fusion enzyme-based in vitro recombination system (Figure 1B). It is predicted that transfection of the recombination reaction mixture into permissive cells could recover the infectious wild type or mutated viruses.
-
The recombination reaction mixture of pJEV-infu and wild type JEV-complement was directly used to transfect BHK-21 cells, and the typical CPE of JEV infection was observed at 5 days p.t. (Figure 2A).
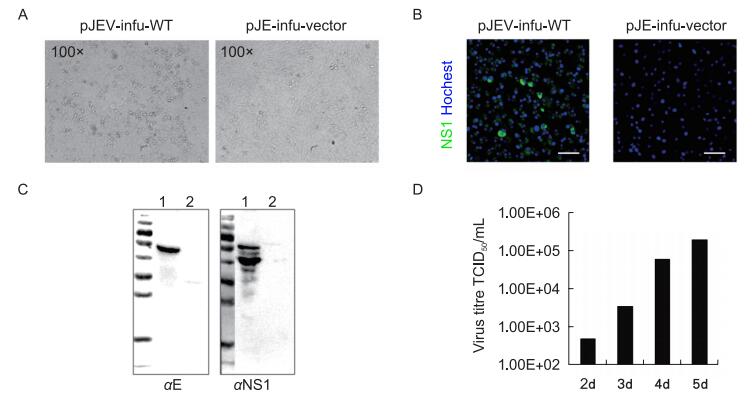
Figure 2. Detection of infectious virus from the pJEV-infu system. (A) The wild-type JEV-complement fragment was cloned into pJEV-infu using the In-Fusion cloning system and the reaction mixture (pJEV-infu-WT) was transfected into BHK-21 cells. Transfection of the linearized pJEV-infu vector was performed in parallel as a negative control. The cytopathic effect was observed at 5 days post transfection. The supernatants of cells transfected with pJEV-infu-WT (1) as well as the pJEV-infu vector only (2) were harvested and used to infect healthy BHK-21 cells. At 36 h post-inoculation, the expression of JEV proteins was detected by an immunofluorescence assay (B) and western blot (C) using the indicated JEV-specific antibodies. Bars indicate 50 μm. (D) The kinetics of wild-type JEV production using the pJEV-infu system after transfection.
To further confirm the generation of infectious viruses with this pJEV-infu system, the supernatant of the transfected cells was used to infect healthy BHK-21 cells. Both IFA and western blot analysis revealed clear expression of JEV proteins (Figure 2B, 2C).
To better understand the efficiency of the pJEV-infu system for virus production, the supernatant of the transfected cells was further analyzed, which revealed that the infectious virus could be released as early as 2 days p.t. Over the course of 5 days, the infectious virus steadily increased to a titer as high as 2 × 105 TCID50/mL (Figure 2D).
-
To determine whether the recovered virus possessed authentic characteristics, the plaque-forming abilities of the viruses were investigated. The virus recovered from the pACYC-JEV system was used as the parental virus for this experiment (Li et al., 2014). As shown in Figure 3A, the virus recovered from the pJEV-infu system produced plaques with a similar size to those of the parental virus.
Figure 3. Phenotypic comparison between the recovered and parental viruses. (A) BHK-21 cells in a 12-well plate were infected with the indicated viruses and incubated under a semi-solid overlay for 3 days. Plaques were visualized by crystal violet staining. (B) BHK-21 cells were infected with the indicated viruses at an M.O.I of 0.01. The virus titer in the supernatants was measured at the indicated times.
Furthermore, the growth curves of both viruses were similar (Figure 3B). This result corroborated the results of the plaque assays, indicating that the virus recovered from the pJEV-infu system was phenotypically identical to the parental virus.
-
The JEV E protein contains 12 cysteines, which are strictly conserved among flaviviruses, thereby highlighting their important contributions to the function of the protein (Luca et al., 2012). To evaluate the role of the cysteines in the JEV E protein, we used the pJEV-infu system to construct a panel of 12 recombinants encoding a single alanine substitution for each of the 12 cysteines of E protein. In parallel, a leucine to phenylanine substitution at amino acid position 107 of the E protein was introduced as a control, since this mutation is predicted to affect virus fusion and thus decrease virus infectivity (Allison et al., 2001).
Mutations were introduced into the structural gene segment (JEV-complement) using OE-PCR with the primers indicated in Table 1, and cloned into pJEV-infu backbone followed by transfection into BHK-21 cells. At 5 days p.t., both the supernatants and transfected cells were harvested separately. The titer of each variant of virus was determined and the relative virus genome copies in transfected cells were quantified by qRT-PCR.
As indicated in Figure 4A, all 12 single cysteine mutations resulted in defective generation of infectious virus. In contrast, the L107F mutation had little effect on infectious virus production except for a slight decrease in the viral titer. Accordingly, the relative number of virus genome copies from cells transfected with the cysteine mutants was at least 104 fold lower than that of cells transfected with the wild-type control, whereas the L107F mutation produced only a 3-fold decrease in genome copy numbers (Figure 4B).
Figure 4. Mutations of the cysteine residues in the envelope (E) protein of Japanese encephalitis virus (JEV) resulted in a defect of generating infectious virus. JEV-complements carrying each cysteine to alanine mutation of E protein were cloned into pJEV-infu respectively and transfected into BHK-21 cells. Transfection of the wild type clone as well as that with a leucine-to-phenylalanine mutation at amino acid position 107 of E protein was performed in parallel as a control. At 5 days post-transfection, (A) the infectious virus in the supernatants was titered and (B) the relative number of viral genome copies in transfected cells was quantified by qRT-PCR. Independent preparations of virus were produced for the C304A and C335A mutants; error bars represent standard errors. (C) The effect of the C304A and C335A mutations on infectious virus production was determined using a previously described pACYC-JEV system (Li et al., 2014). The indicated mutations were introduced into the pACYC-JEV plasmid, and viral RNA carrying the corresponding mutations was obtained via in vitro transcription. A total of 200 ng each RNA was transfected into BHK-21 cells in a 24-well plate, and the expression of JEV proteins was detected by an immunofluorescence assay at 48 h post-transfection. Transfection of wild-type virus RNA transcripted from pACYCJEV was performed as a control. Bars indicate 50 μm.
As a proof-of-principle, the effect of two mutations, C304A and C335A, were validated by previously described pACYC-JEV system (Li et al., 2014). The mutated virus RNAs were generated and transfected into BHK-21 cells, which were stained with JEV-specific antibodies at 48 h p.t. As shown in Figure 4C, no obvious amplification of the mutated viruses could be observed, whereas the wild type virus spread quickly. This result further confirmed that mutation of the cysteines in E protein led to failure in the generation infectious viruses.
Construction of a simple reverse genetic system of JEV
Characterization of the pJEV-infu system
Phenotype characterization of the recovered virus
Rapid mutagenesis of the conserved cysteines in E protein
-
In this study, we established a reverse genetic system of JEV that can rapidly produce infectious virus. Moreover, this approach could be used to genetically modify the structural proteins of JEV at a large scale.
Recently, the genome instability of bacteria has become a major obstacle in constructing infectious clones of JEV. To circumvent this problem, we modified an in vitro ligation method that has been used to construct a molecular clone system of WNV (Lin et al., 2012). By deleting the structural genes in the unstable region, a truncated form of JEV was constructed under the transcriptional control of the CMV promoter to generate the backbone plasmid pJEV-infu. A sequence corresponding to the deleted region, which could be easily genetically modified, was then cloned into pJEV-infu by In-Fusion enzyme-based in vitro recombination. Transfecting the recombination reaction mixture directly into permissive cells allowed for the rapid generation of infectious virus. Compared to the endonuclease-based in vitro ligation method, which usually requires introduction of a unique endonuclease site by synonymous mutations, the In-Fusion enzyme-based in vitro recombination method could ensure construction of the authentic sequence of the virus genome without any undesired modification.
Applications of CMV promoter-controlled in vivo transcription of the JEV genome have been well investigated, and the recovered virus has been confirmed to possess an authentic 5′-end (Yamshchikov et al., 2001). Further addition of HDVr at the 3′-terminus of the genome could also ensure correct processing of the 3′-end (Li et al., 2014). Our data showed that the recovered virus produced plaques of similar size with an identical growth curve to the parental virus, which demonstrated the success and potential of this pJEV-infu system.
Because of the ease of genetic manipulation and its timesaving advantage, this approach could be applied in studies investigating many different aspects of JEV infection. In this study, we used the pJEV-infu system to evaluate the role of the strictly conserved cysteines in E protein throughout the virus life cycle. No infectious progeny virus could be recovered from any molecular clone carrying cysteine-to-alanine mutations. Both the wild type clone and L107F E protein mutant could otherwise generate the infectious virus successfully, although the titer of the virus with the L107F mutation was slightly lower than that of the wild type virus. These results suggested that all of the cysteines of E protein are very important for the virus life cycle.
The 12 cysteines of E protein form six intramolecular disulfide bonds, which have been shown to contribute to the function and antigenicity of many viral glycoproteins (Fenouillet et al., 2008). Moreover, disulfide bridging and/or isomerization play an essential role during protein folding, virus assembly, and infectivity (Carleton and Brown, 1996; Whitehurst et al., 2007; Parrott et al., 2009). Accordingly, mutation of the cysteines of E protein might interfere with maturation of the protein, virus assembly, or infectivity, all of which might result in a defect in infectious virus generation.
To summarize, we circumvented the instability of the JEV genome in bacteria and established a reverse genetic system for rapid generation of infectious virus by using the newly developed In-Fusion enzyme-based in vitro recombination method. Using this approach, we confirmed the key role of the conserved cysteines in E protein during the JEV life cycle. This approach should facilitate further fundamental/basic research on the structural proteins of JEV, including their properties in recognition of neutralizing antibodies, receptor binding, virulence determination and host adaptation, and so on. Moreover, this approach has significant potential for clinical applications, such as vaccine development by substitution of virulence-related amino acids.
-
This work was supported by grants from the National Science Foundation of China (NSFC) (No. 31125003 and No. 31321001) and the National Basic Research Program (973 Program) of China (Grants 2010CB530103). We thank Professor Bo Zhang for kindly providing the infectious clone plasmid JEV pACYC-JEV. We acknowledge the Core Facility and Technical Support of Wuhan Institute of Virology for technical assistance.
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
RD performed the experiments. RD, FD and HW conceived the study and participated in the design. MW and ZH provided experimental support. RD, FD drafted the manuscript. All authors read and approved the final manuscript.

 DownLoad:
DownLoad: