












HTML
-
Before the outbreak of the severe acute respiratory syndrome(SARS)epidemic worldwide in the winter of 2002 to the summer of 2003, coronaviruses(CoVs)had long been considered to cause merely mild symptoms in humans despite being attributed to severe diseases in animals(Dominguez et al., 2003; Drosten et al., 2003; Franco-Paredes et al., 2003; Kuiken et al., 2003; Rota et al., 2003). The SARS epidemic, caused by the SARS coronavirus(SARS-CoV), resulted in a total of 775 deaths out of 8, 273 cases of infection(Dominguez et al., 2003; Franco-Paredes et al., 2003). In 2012, another new CoV called the Middle East respiratory syndrome corona-virus(MERS-CoV)emerged, which originated from the Middle East(Zaki et al., 2012). According to the World Health Organization, this new CoV has thus far resulted in 1, 633 cases of infection causing 587 deaths(http://www.who.int/emergencies/mers-cov/en/). Both SARS-and MERS-CoVs have zoonotic origins(Woo et al., 2009; Woo et al., 2012; Chan et al., 2013; Ge et al., 2013; Lu et al., 2015; St John et al., 2015), strongly suggesting that animal CoVs remain a potential threat to humans. However, no specific drugs have been approved to treat CoV-associated diseases to date.
In the wake of the SARS outbreak, in-depth research has revealed multiple aspects of the life cycle of CoV infection. CoVs mainly take advantage of spike proteins to bind their receptors for attachment to the host cell membrane(Ng et al., 2003). Upon entering host cells, CoVs start to release genomic RNA. Subsequent expression of two polyproteins responsible for viral replication and transcription, respectively, requires hijacking of host ribosomes to facilitate translation(Thiel et al., 2003). The two resulting polyproteins are further processed by two viral-encoded proteases into 16 mature nonstructural proteins(nsps): Nsp1, a host gene expression suppressor; nsp2, which functions in interacting with the two host proteins; nsp3, a multi-domain complex; nsp4/nsp6, transmembrane proteins; nsp5, the Mpro; nsp7/nsp8, primases; nsp9, a single-stranded RNA-binding protein; nsp12, an RNA-dependent RNA polymerase; nsp13, 5′-to-3′ RNA helicase/NTPase/RNA 5′-triphosphatase; nsp14, 3′-to-5′ exoribonuclease; nsp15, uridylate-specific endoribonuclease; and nsp16, 2′-O-methyltransferase(Cornillez-TY et al., 2009; te Velthuis et al., 2012; Ivanov et al., 2004; Narayanan et al., 2008; Snijder et al., 2003; Sutton et al., 2004; Ziebuhr, 2005). These nsps can then assemble into the replication-transcription complex and facilitate viral RNA replication and transcription(Zhao et al., 2013a). Currently, several viral proteins are considered candidate targets for drug development, including proteases, helicases, RdRp, and methyltransferase, which are all involved in the aforementioned viral life cycle(Adedeji and Sarafianos, 2014). Among them, Mpro is an important target, owing to its essential role in the processing of polyproteins(Ziebuhr et al., 2000). In this review, we will focus on recent progress in the development of inhibitors of these proteases, with a particular focus on Mpro(Table 1).
Targets Inhibitors Effective/inhibitory concentration References PLpro 5c 9.5 μmol/L(EC50) Baez-Santos et al., 2014 Mpro 8k1 1.04 μmol/L(IC50) Liu et al., 2014; decahydroisoquinolin scaffold 41 63 μmol/L(IC50) Shimamoto et al., 2015 Cbz-TSAVLQ-CN 1.3-4.6 μmol/L(IC50) Chuck et al., 2014 NPI52 0.02 μmol/L(EC50) Kim et al., 2015 Note: EC50: the half maximal effective concentration; IC50: the half maximal inhibitory concentration. Table 1. Inhibitors targeting papain-like protease (PLPro) and main protease (Mpro).
-
Papain-like protease(PLpro)(Figure 1A)exists as a functional domain within the large nsp3, which also encodes a ubiquitin-like fold, an ADP-ribose-1d-phosphatase domain, a SARS-CoV-unique domain, and a transmembrane domain(Lee et al., 2015). PLpro recognizes a specific site comprising the consensus cleavage sequence LXGG between nsp1/2, nsp2/3 and nsp3/4(Lee et al., 2015). Proteolytic cleavage of the peptide bond after the glycine residue can result in the release of nsp1, nsp2, and nsp3 from the viral polyproteins, which is a critical step for viral replication(Ziebuhr et al., 2000).
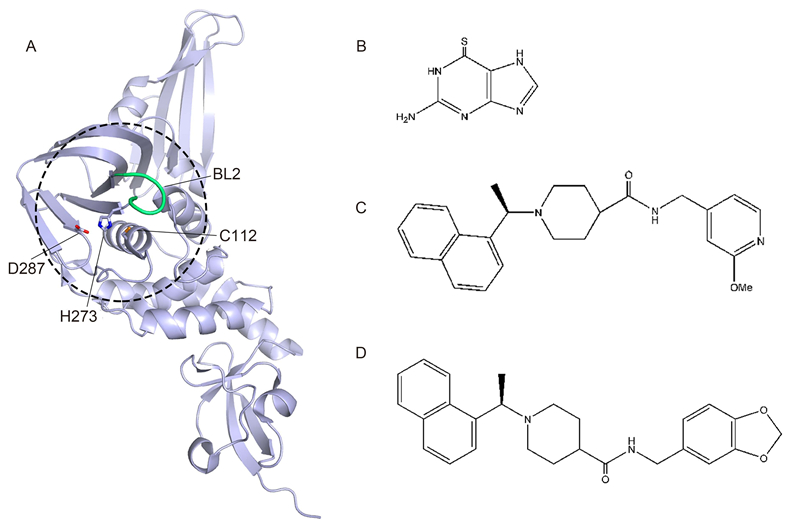
Figure 1. The structures of CoV PLpro, its inhibitors, and derivatives. (A) Overall structure of SARS-CoV PLpro (PDB entry 2FE8). The hotspot for inhibitor development is depicted by the dashed circle, with the active site residues (C112, H273, D287) and BL2 loop shown as sticks and a tube, respectively. (B) 6-Thioguanine compound. (C) Methoxypyridine inhibitor 5c. (D) PLpro inhibitor GRL-0667.
PLpro has been found to have deubiquitination and de-interferon stimulated gene-ylation(ISGylation)functions(Chen et al., 2015). This can help the virus to escape the host immune response via the interferon signaling pathway(Baez-Santos et al., 2015). In MERS-CoV, PLpro has been reported to protect polyproteins from cleavage through the interferon regulatory factor 3 pathway(Sun et al., 2012). PLpros from different CoV species may also vary in substrate specificity(Lee et al., 2015), which may contribute to the difficulty in developing a wide-spectrum drug targeting CoVs. Nonetheless, some molecules have been found to effectively inhibit CoV infection, including zinc ion(Zn2+) and zinc conjugate inhibitors, which were reported to target PLpro(Han et al., 2005). However, these inhibitors appear to be unique, because no other metal ions have thus far been found to possess such inhibitory capacity(Baez-Santos et al., 2015). In addition, thiopurine compounds could suppress the activity of PLpros by covalently binding to the cysteine residue at the active site(Chen et al., 2009). Some natural products have also been found to inhibit PLpro activity, including tanshinones(Park et al., 2012b), diarylheptanoids(Park et al., 2012a), and geranylated flavonoids(Cho et al., 2013).
Recently, several crystal structures of SARS-CoV PLpro in complex with inhibitors have been reported(Ratia et al., 2008; Ghosh et al., 2009; Ghosh et al., 2010; Baez-Santos et al., 2014)(Figure 1A-1D). SARS-CoVPLpro contains two blocking loops, BL1 and BL2, which have proven to be vital in blocking access to the catalytic site(Lee et al., 2015). Upon binding to the BL2 loop, these inhibitors cause a local conformational change and subsequently block access for substrate binding to the catalytic site(Lee et al., 2015).
-
The CoV genome encodes two overlapping polyproteins designated as pp1a and pp1ab(Mukherjee et al., 2008). The polyproteins are cleaved by viral proteases to yield the 16 mature nsps mentioned above, and assemble into the replication-transcription complex that is essential for viral replication. Mpro is the key enzyme involved in this proteolysis process(Ziebuhr et al., 2000), and is responsible for cleavage at 11 sites on the polyproteins(Ziebuhr, 2004). Owing to its dominant and indispensable role in polyprotein processing, Mpro has thus emerged as an important target for anti-CoV drug design.
Many of the inhibitors identified to date, such as peptidomimetic(Ghosh et al., 2007), TG-0205221(Yang et al., 2006), ketones(Shao et al., 2008), and pyrimidines(Ramajayam et al., 2010), have previously been reviewed in detail(Mukherjee et al., 2011; Zhao et al., 2013a; Zhao et al., 2013b; Adedeji and Sarafianos, 2014). Therefore, we here focus only on the most recent progress in inhibitor and drug discovery.
In the last two years, many research groups have exerted continuous effort on the development of inhibitors targeting CoV Mpros(Figure 2A). Chen et al. and Zhou et al. reported a new class of isatin derivatives as Mpro inhibitors(Chen et al., 2005; Zhou et al., 2006). Based on their results, Liu et al. started to investigate the potential of using isatin derivatives to design more efficient inhibitors against CoV Mpros(Liu et al., 2014). In these studies, a series of isatin derivatives were designed, synthesized, and assessed by in vitro enzymatic assays with a fluorogenic substrate peptide. Among them, the 5-sulfonyl isatin derivatives with a carboxamide group substituted by a series of sulfonamide groups showed inhibition against Mpro at the micromolar range. Assisted by a structure-based lead compound design, the authors effectively optimized the 5-sulfonyl isatin derivatives and obtained an optimal compound named 8k1 (IC50 = 1.04 mol/L)(Figure 2B). By comparisons of different substituted groups, they reached the conclusion that 5-sulfonyl isatin coupled with methyl at the N-1 position could dramatically promote inhibitory activity, which could facilitate further lead compound development.

Figure 2. The structures of CoV Mpro, its inhibitors, and derivatives. (A) Overall structure of SARS-CoV Mpro (PDB entry 1UK3). Two protomers are shown in lime green and light blue, respectively. The substrate-binding pocket is shown in yellow in the inset, with subsites labeled. (B) Compound 8k1 of isatin derivatives. (C) Inhibitor derived from the decahydroisoquinolin scaffold. (D) An irreversible compound N3. (E) Peptidyl compound NPI52.
Akaji's group reported a novel inhibitor with a decahydroisoquinolin scaffold that could target SARS-CoV Mpro(Shimamoto et al., 2015)(Figure 2C). Previous studies showed that the hydrophobic interactions at the S2 site are important for substrate/inhibitor binding. This new inhibitor was designed by connecting the substrate-based inhibitor's cyclohexyl group, located at the P2 site, with the -nitrogen atom of the P2 position on the main chain. This approach could maintain the hydrophobic interactions at the S1 and S2 sites, which kept the imidazole and aldehyde groups in the required configuration for respective binding. The authors further synthesized a series of decahydroisoquinolin derivatives, all of which showed moderate but clear inhibitory activities against SARS-CoV Mpro. X-ray crystallographic studies revealed that the decahydroisoquinolin scaffold was inserted into a large S2 pocket, which occupied most of the pocket to hold the terminal aldehyde tightly inside the active site cleft, resulting in the observed inhibitory activity.
Previously, our group investigated the conservation of CoV Mpros across species. Structural analyses revealed that the substrate-binding pockets of various CoV Mpros are highly conserved, which led to the concept of "wide-spectrum inhibitors" for targeting all CoVs. Through a structure-based drug design, we have identified a lead compound named N3 with potent inhibitory activity against all Mpros tested(Figure 2D). Recently, the Wong group analyzed the substrate specificity of Mpro from the P5 to P3' sites among several CoVs, and found that all Mpros have similar substrate specificity. This finding facilitated the design of a broad-spectrum peptidomimetic inhibitor against various CoV Mpros. They identified a new peptidyl compound, Cbz-TSAVLQ-CN, by coupling a nitrile warhead and a carboxybenzyl(Cbz)group to the C-terminus and N-terminus of a peptide, respectively(Chuck et al., 2014). Enzymatic assays revealed that it could inhibit Mpros from six different species of CoVs. The solved crystal structure of the enzyme in complex with this inhibitor indicated that the N-terminal Cbz protective group could improve the interaction between the inhibitor and the protease. Kim et al. also designed a series of peptidyl compounds targeting feline coronaviruses(FCoV) and feline caliciviruses(FCV)(Kim et al., 2015). They reported that a compound named NPI52 displayed inhibitory activity against FCoV and FCV strains in cell culture(Figure 2E). This inhibitory effect was further confirmed in a mouse model based on liver histopathology analysis.
In addition, we have designed N3 and zinc ion as dual inhibitors against feline infectious peritonitis virus(FIPV) Mpro. The biochemical data showed that N3 and Zn2+ could serve as an irreversible inhibitor and a non-competitive inhibitor, respectively. The solved crystal structure of FIPV Mpro in complex with these two inhibitors demonstrated their potential for synergistic activity to achieve an enhanced inhibitory effect(Wang et al., 2015). These findings provide a new approach in the design of anti-CoV drugs.
On the other hand, gaining a better underst and ing of the cleavage mechanism of CoV Mpros could help to provide new directions for the design of inhibitors. Dimerization is an essential step in Mpro maturation for subsequent catalysis. Shi et al. reported the mechanism of CoV Mpro for controlling the dimer-monomer switch(Shi et al., 2008). Specifically, mutation of the residue Arg298 at the dimer interface was shown to disturb the dimerization of Mpro (Paasche et al., 2014), which could be reverted via a substrate-induced conformational change. Moreover, structural studies revealed that the establishment of a new intermolecular interaction required domain III composed of a globular cluster of five helices(Anand et al., 2003)to alter its orientation. This suggests that Mpro should be able to tolerate large conformational changes without loss of enzymatic activity. This further implies that intramolecular communication and regulation exists within the Mpro structure. Thus, better underst and ing of the underlying mechanism might aid in structure-based anti-CoV drug design.
Paasche et al. reported substrate binding-induced zwitterion formation at the active site of SARS-CoV Mpro (Paasche et al., 2014). They discovered that the Cys-/His+ zwitterionic state is essential for efficient proteolytic activity, which is fostered by substrate binding rather than by inhibitor binding, and enhances its substrate specificity. The free-energy calculations delineated that substrate binding could promote the proton transfer reaction, which reduces the energy between neutral and zwitterionic states. This implies that development of antiviral agents with ability of the substrate to trigger the zwitterionic state may be an alternative approach for drug design.
Papain-like protease
Main protease
-
In the wake of outbreaks of SARS and MERS, CoVs have emerged as life-threatening pathogens for humans. Their zoonotic origin also sends a warning of the potential emergence of new deadly CoVs via interspecies transmission. Thus, many inhibitors have been developed to prevent CoV infection. However, the problems of toxicity, ability to enter cells, non-specificity, and instability(particularly for peptide-derived inhibitors), among others, are still major hurdles to overcome for these lead compounds. Therefore, further investigation is required to develop an effective treatment to prevent CoV infection, which will require tackling the problem with multiple approaches.
-
This work was supported by the Tianjin University Undergraduate Research Foundation, Tianjin University-Hainan University Collaborative Foundation, National Natural Science Foundation of China(No. 31300150), and Tianjin Marine Science and Technology Program(No. KJXH2014-16).
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: