









-
Bats are natural reservoirs of many viruses; some severely affect other mammals and humans (O’Shea et al., 2014). These diseases include those caused by the Ebola virus, Nipah virus, lyssaviruses, Japanese encephalitis virus, coronaviruses and Hendra virus (Calisher et al., 2006; Liu et al., 2013; Dietrich et al., 2015; Zheng et al., 2016). Bats carry many pathogens but seldom display pathogenic symptoms, which is of great interest to scientists who study bat viruses and virus-host interactions. Considering the diversity of the bat population, it is expected that there are more bat viruses waiting to be discovered (Shi, 2013).
Because Macau is a globalization metropolis, it is necessary to develop surveillance against zoonotic viruses for the early warning of potential emerging infectious diseases. There was evidence that epidemic diseases broke out in Macau. First example, there was an epidemic fever caused by dengue and dengue-like viruses in 1874 (Buchillet, 2012), and in 1932, an outbreak of cere- brospinal meningitis caused 491 infectious cases and 290 deaths (Buchillet and de Lamballerie, 2010). A relatively recent epidemic event occurred in 2009. Infected patients were identified as infected by the pandemic influenza A virus H1N1 (Zhou et al., 2010). However, the prevalence and distribution of bat viruses in Macau was unknown before this study. In this study, we performed an investigation of virus infection in bat populations in Macau from 2015 to 2016.
-
From April 2015 to March of 2016, bat samples were collected from 7 locations (Table 1 and Figure 1). Bat species were identified by morphology and further classified by cytochrome b amplification and sequencing. For fecal samples, clean plastic sheets were laid down on flat surfaces beneath bats roosts before sunset (Ge et al., 2012), and samples were collected the next day. To collect anal swabs, individual bats were randomly caught, and each individual was gently swabbed once or twice on their anal verge. Fecal pellets and anal swabs were preserved in 1 mL virus transportation medium (VTM), placed into liquid nitrogen and stored at –80 °C in the laboratory before use.
Bat species Pattern No. of tested samples (positive) Location Time (month/year) swab/faces/urine (CoV)(AdV)(PMV) Familiy Pteropodidae Cynopterus sphinx 1/0/0 1(0)(0)(0) 4 3/2016 Eonycteris spelaea 0/1/0 1(0)(0)(0) 2 5/2015 Familiy Rhinolophidae Rhinolophus pusillus 0/356/0 356(2)(0)(1) 2, 5 4, 5, 9, 10, 12/2015; 2/2016 Rhinolophus sinicus 0/0/2 2(0)(0)(0) 6 11/2015 Familiy Hipposideridae Hipposideros armiger 0/421/0 421(0)(4)(0) 2, 4, 6, 7 4, 6, 7, 8, 9, 10, 12/2015; 3/2016 Famliy Vespertilionidae Pipistrellus abramus 8/4/0 12(0)(2)(0) 3 4, 9/2015 Pipistrellus minus 13/0/0 13(0)(0)(0) 3 9/2015 Miniopterus schreibersii 78/0/0 78(24)(0)(1) 1 7/2015; 3/2016 Myotis ricketti 42/0/0 42(10)(16)(0) 1 7/2015; 3/2016 Tylonycteris pachypus 78/0/0 78(14)(0)(0) 4 4, 7/2015; 5/2016 Table 1. Samples collected and virus detection results
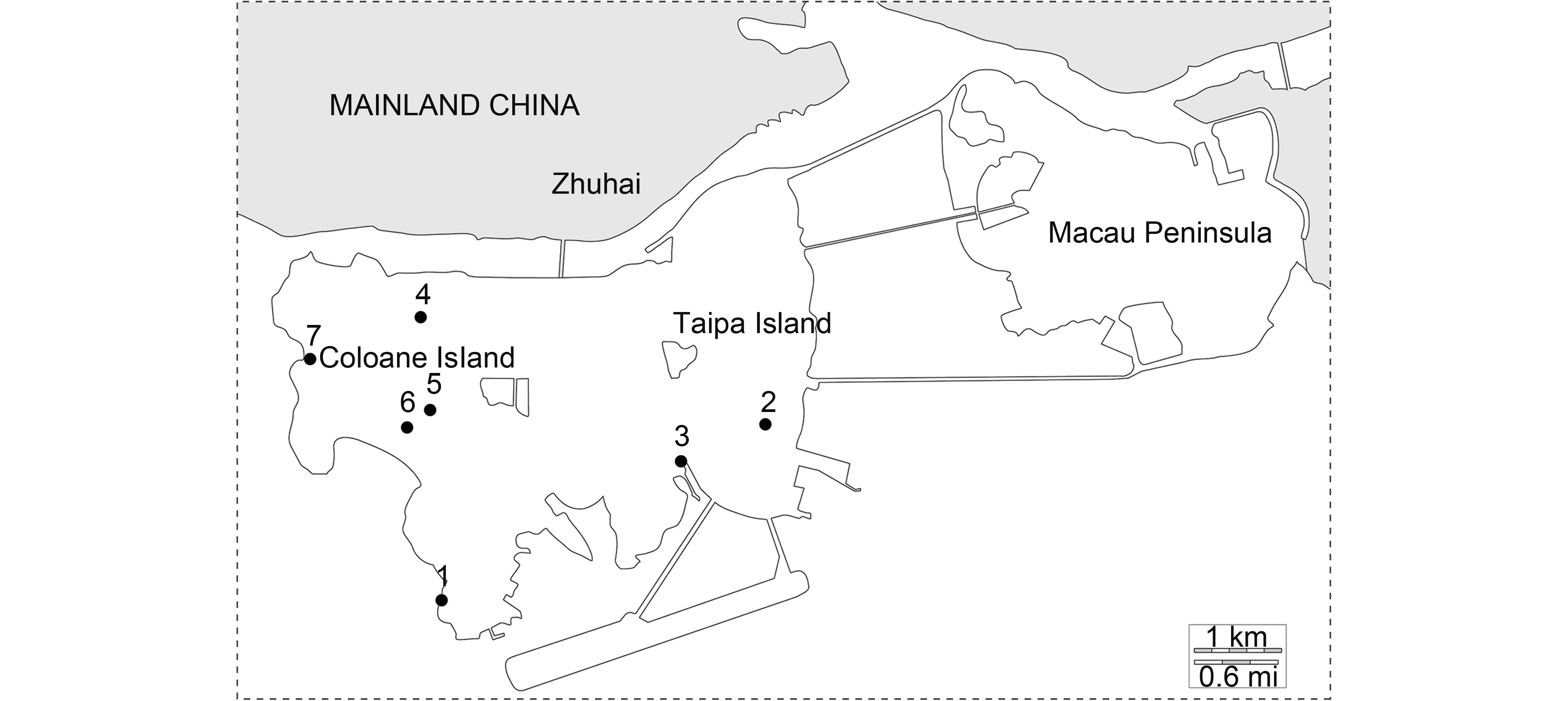
Figure 1. Map showing bat surveillance sites in Macau. Areas belonging to Zhuhai of Mainland China are shaded. The solid dots represent 7 locations where bat samples were collected.
-
Total nucleic acid was extracted using an RNA Extraction Kit (Roche, High Pure Viral RNA kit) according to the manufacturer’s instructions. Previously published methods were used to detect coronavirus, adenovirus (AdV) and paramyxovirus (PMV). For coronavirus, samples were screened for the presence of virus using modified nested RT-PCR with primers targeted against the RNA dependent RNA polymerase (RdRp) (Poon et al., 2005). For adenovirus, samples were screened by nested-PCR using degenerate primers based on the conserved domain of the polymerase of adenovirus genomes (Li et al., 2010). For paramyxovirus, a hemi-nested RT-PCR targeting the L gene was performed as described previously (Tong et al., 2008). For hepatitis A virus, hepatitis E virus and hantavirus, the detection methods were performed as previously described (Klempa et al., 2006; Drexler et al., 2012; Drexler et al., 2015). The amplicons were sequenced directly or cloned into the pGEM-T Easy vector (Promega) if direct sequencing failed.
-
The obtained sequences were aligned with homologous sequences downloaded from NCBI using ClustalW. Phylogenetic trees were constructed with MEGA7 using maximum likelihood analyses. Evaluation of the statistical confidence of each node was based on 1000 bootstrap replicates (Chen et al., 2016).
-
Virus isolation was performed in the biosafety level (BSL)-2 facility at Wuhan Institute of Virology, Chinese Academy of Sciences. Positive samples were vortex blended before centrifugation at 1000 × g for 5 min. The supernatant was clarified and inoculated onto VERO cells. The HKU4 positive samples were incubated on hepatocellular carcinoma huh cells (Huh7) and pneumocyte cells of Tylonycteris pachypus collected in Guangzhou. After twenty serial passages, RNA samples were extracted from each culture supernatant and analyzed by nested RT-PCR to confirm the success of virus isolation (Sasaki et al., 2012).
-
A total of 220 anal swabs, 782 fecal pellets and 2 urine samples were collected from 10 bat species (Table 1). Of the 1004 samples, 50 were positive for coronaviruses, 22 for adenoviruses and 2 for paramyxoviruses among 6 bat species. We also selected 207 samples from 6 species to detect hepatitis A virus (HAV), hepatitis E virus (HEV) and hantavirus (HTNV), but none was positive. The positive rate of PCR in location 1 was higher than in other locations (Figure 1). Cohabitation of two bat species was observed in several locations. For example, Miniopterus schreibersii and Myotis ricketti were found living in the same cave in location 1, Rhinolophus pusillus and Hipposideros armiger in location 2, Pipistrellus abramus and Pipistrellus minus in location 3 (Figure 1).
Interestingly, coronavirus and adenovirus had a high rate of co-infection in Myotis ricketti, and all M. ricketti infected with HKU6 were also infected by adenovirus (Table 2). However, no virus isolation was successful.
Virus species Bat species and number of individuals in which viruses were detected R. pusillus H. armiger P. abramus M. schreibersii M. ricketti T. pachypus CoV HKU4 14 HKU7 1 1A/1B 2 23 HKU6 10 AdV Unclassified 4 2 16 PMV Unclassified 1 1 Co-infection 1A/Unclassified PMV 1 HKU6/Unclassified AdV 10 Note: Abbreviated species names: R. pusillus, Rhinolophus pusillus; H. armiger, Hipposideros armiger; P. abramus, Pipistrellus abramus; M. schreibersii, Miniopterus schreibersii; M. ricketti, Myotis ricketti; T. pachypus, Tylonycteris pachypus. Table 2. Coronaviruses, adenoviruses and paramyxoviruses detected in different bat species
-
Coronaviruses were detected in 50 of 1004 samples, including R. pusillus (2/356, 0.56%), M. schreibersii (24/78, 30.77%), M. ricketti (10/42, 23.81%) and T. pachypus (14/78, 17.95%) (Table 1). The partial RdRp gene sequences (360 bp) were obtained from 387 bp PCR amplicons and were used for phylogenetic analysis (KY783855-KY783903) (Figure 2). These sequences were divided into two categories, Alphacoronavirus (αCoV) and Betacoronavirus (βCoV), with a large proportion corresponding to αCoV. High CoV infection frequencies were found in M. schreibersii, M. ricketti, T. pachypus and R. pusillus. Of the 36 samples that were positive for Alphacoronavirus, 24 were from M. schreibersii and were related to BtCoV 1A (18), BtCoV 1B (5) or BtCoV HKU7 (1); 2 were from R. pusillus and were related to BtCoV 1 and BtCoV 1A; and 10 were from M. ricketti and were related to BtCoV HKU6 (Figure 2). Most sequences shared > 92% nucleotide (nt) identity with the closest reference sequence of known viral species. Fourteen viruses detected in this study belonged to Betacoronavirus HKU4, and all were from T. pachypus. These HKU4-like viruses share > 96% nt identity with two strains of HKU4-related viruses (BtTp-BetaCoV/ GX2012/KJ473822.1 and BtCoV/242/2005.DQ648803.1).
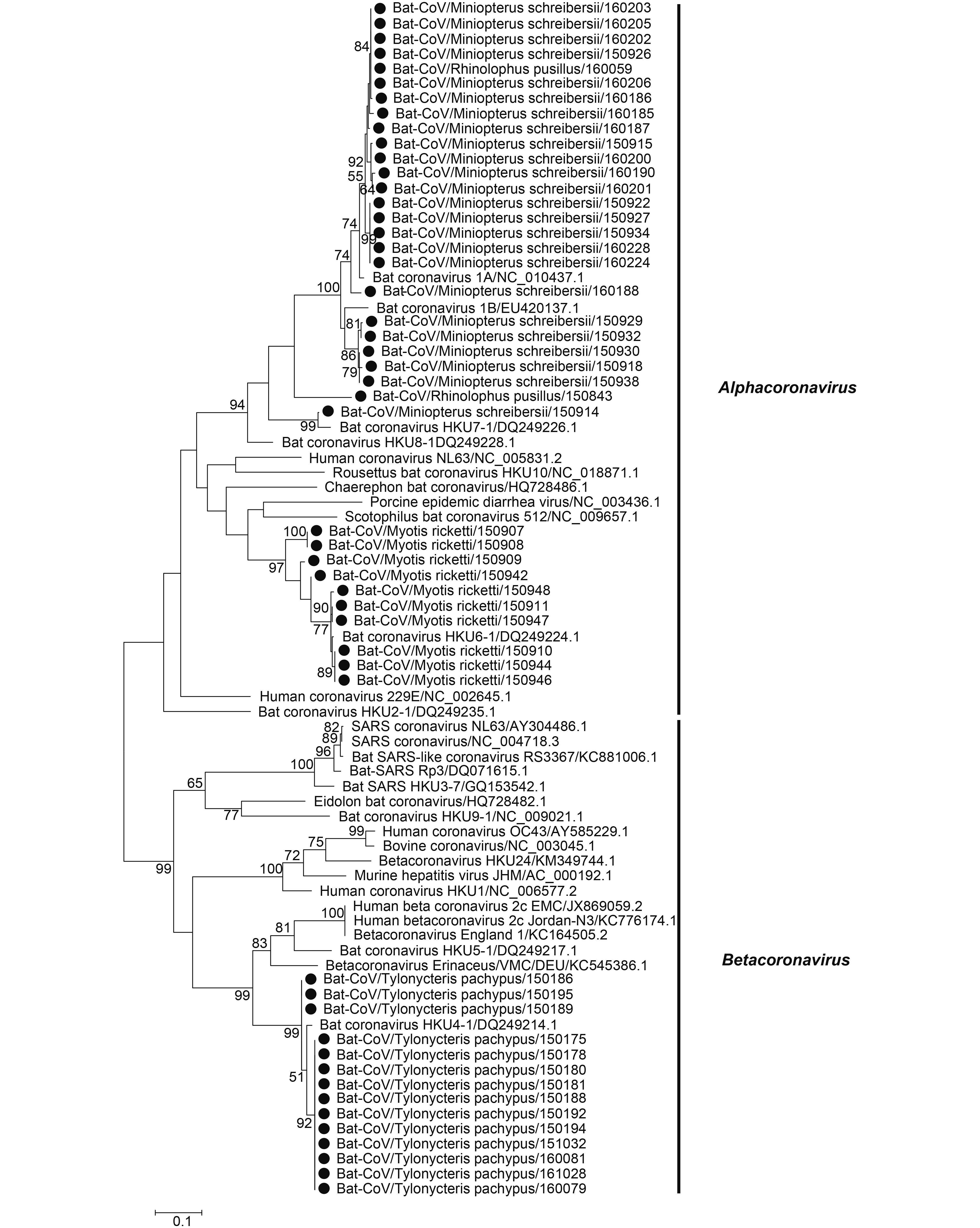
Figure 2. Phylogenetic construction based on the partial (360 bp) RdRp gene sequences of coronavirus. Viruses detected in this study are highlighted by a solid circle. The tree was constructed using the maximum-likelihood method with a 1000 bootstrap test using MEGA7.0.
-
Twenty-two samples were positive for adenovirus and all were detected in the following three species: P. abramus (2/12, 16.6%), M. ricketti (16/42, 38.10%) and H. armiger (4/422, 0.95%). No positive results were found in other species. The partial DNA-dependent DNA polymerase gene (Pol) sequences from the same bat species shared high identity. One bat from each of the three bat species was used for phylogenetic analysis based on translated amino sequences (KY783852-7K783854) (Figure 3). The adenoviruses were divided into 4 groups, Aviadenovirus, Siadenovirus, Atadenovirus, and Mastadenovirus, according to a previously published study (Tan et al., 2016). All tested sequences in this study belong to the Mastadenovirus group. BLAST analysis showed that AdVs from H. armiger possessed > 96% nt identity to Bat-AdV 1391-YN-Ha (GU226964.1), those from M. ricketti possessed > 97% nt identity to Bat-AdV 1285-YN-Mr (GU226966.1), and those from P. abramus possessed 78% nt identity to lesser noctule adenovirus 3 strain 239/08 (KM043105.1).
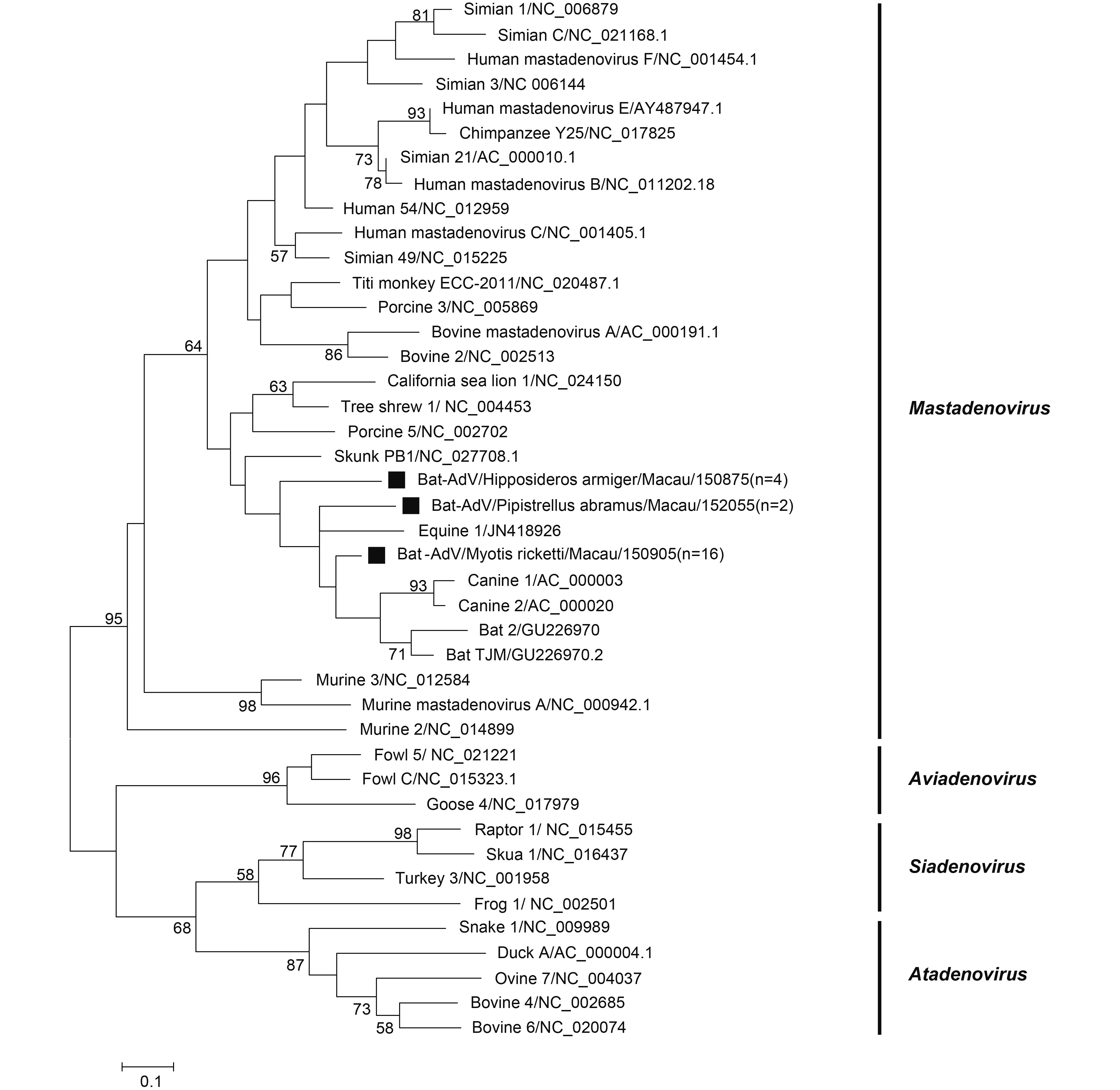
Figure 3. Phylogenetic construction based on translated amino acid sequences from 260 bp partial Pol gene of adenovirus. Viruses detected in this study are highlighted by a solid square. The tree was constructed using the maximum-likelihood method with a 1000 bootstrap test using MEGA7.0.
-
Only two samples were positive for paramyxovirus: one was from M. schreibersii (1/78, 1.28%), and the other was from R. pusillus (1/356, 0.28%). A phylogenetic tree was constructed based on the 531 bp partial L gene of members of Paramyxoviridae family. In the phylogenetic tree, paramyxoviruses were divided into 9 groups (Maganga et al., 2014). The two viruses detected in this study were clustered with the unclassified paramyxoviruses (KY783904-KY783905) (Figure 4). BLAST analysis showed that both shared 71% nt identity to J-virus; one of them shared 91% nt identity with its closest relative, Bat-PV/YN12103/CHN/2012 (KC599258.1); and the other shared 78% nt identity with Bat-PV/ Anhui2011 (KC154054.1)
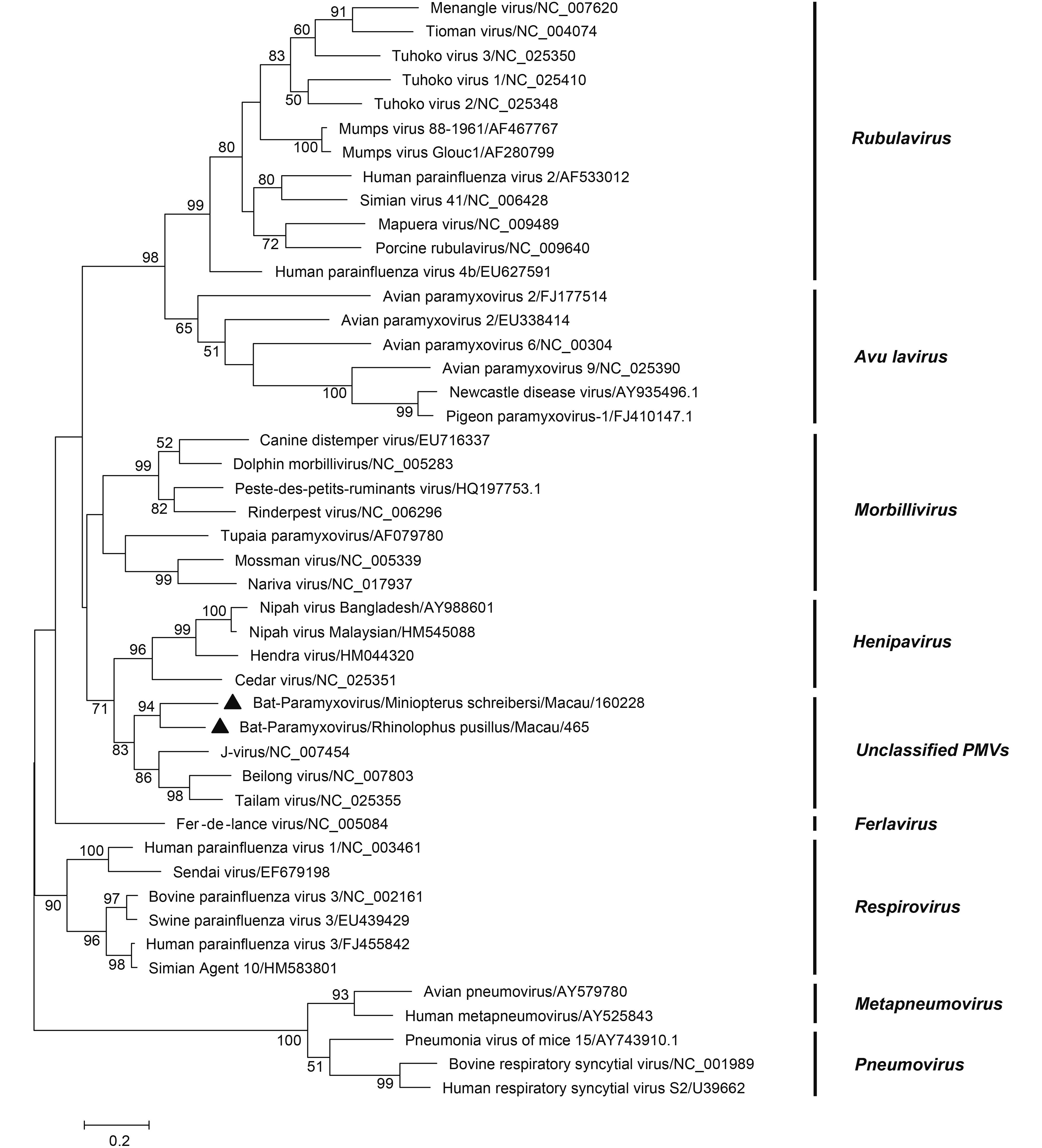
Figure 4. Phylogenetic construction based on the partial L gene (531 bp) sequences of paramyxovirus. Viruses detected in this study are highlighted by a solid triangle. The tree was constructed using the maximum-likelihood method with a 1000 bootstrap test using MEGA7.0.
-
To further address potential cross-species infections of coronavirus found in the study, we randomly selected several HKU4- and 1A-related samples to amplify the full-length S gene. Seven full-length S genes from BtCoV 1A-related samples (KY783845-KY783851) and three from BtCoV HKU4-related samples (KY783842-KY783844) were successfully amplified. Phylogenetic analysis based on these full-length S genes show that among the Alphacoronavirus, four strains from M. schreibersii and one from R. pusillus clustered together and possessed 91% to 93% nucleotide identity with BtCoV 1A (Figure 5). Two other strains from M. schreibersii were clustered with BtCoV/AH2011/KJ473795 and BtCoV/JX2012/KJ473796, both sharing 89% nt identity with BtCoV 1A. The seven BtCoV1A-related S genes shared 88.1% to 100% nt identity. The three strains from T. pachypus related to HKU4 were clustered with BtCoV/ 133/2005/DQ648794 (Figure 5). Sequence comparison showed that the S gene of the three HKU4-related strains shared 97.9% to 99% nt identity among themselves and shared 95.3% to 96.1% nt identity with BtCoV HKU4 (NC_009019), 68.8% to 69.1% nt identity with BtCoV HKU5 (NC_009020), and 67.4% to 67.8% nt identity with MERS-CoV (NC_019843) (Table 3). BtCoV HKU4 has been demonstrated to use dipeptidyl peptidase 4 (DPP4) as its receptor for cell entry, similarly to MERS-CoV (Yang et al., 2014) . The potential threat of the HKU4-like bat coronaviruses to public health cannot be underestimated as their spike may adapt to human cells for cross-species infection.
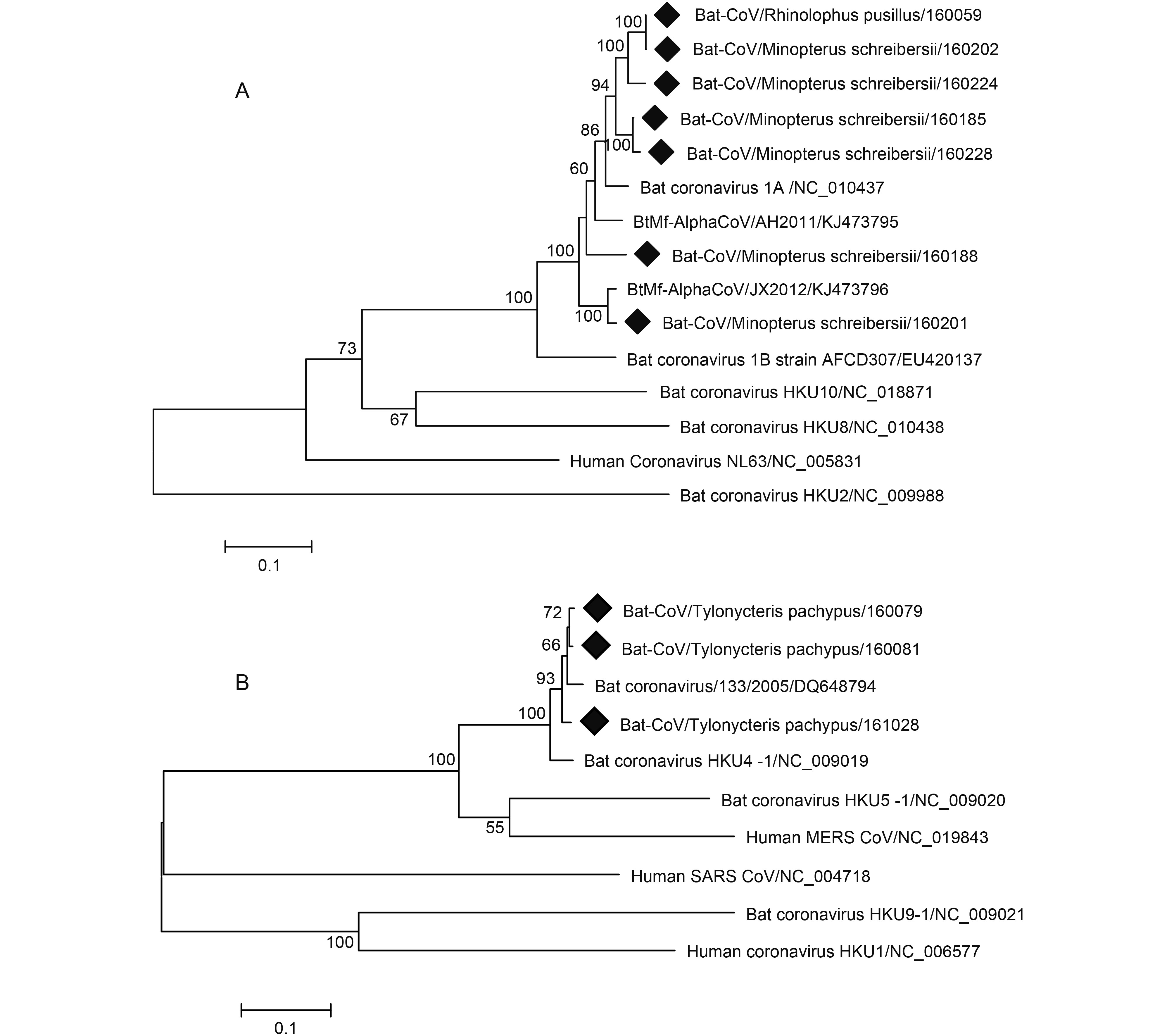
Figure 5. Phylogenetic construction based on the complete S gene sequences of coronavirus. Viruses detected in this study are highlighted by a solid diamond. The tree was constructed using the maximum-likelihood method with a 1000 bootstrap test using MEGA7.0. (A) Alphacoronavirus. (B) Betacoronavirus.
Strain Accession No. Host species BtTpCoV160079 BtTpCoV160081 BtTpCoV161028 Nucleotide sequence identity/Amino acid sequence identity 133/2005 DQ648794 Tylonycteris pachypus 97.4/97.6 97.4/97.6 93.6/97.9 GX2012 KJ473822 Tylonycteris pachypus 93.2/96.2 93.2/96.2 96.3/96.3 NeoCoV KC869678 Neoromicia capensis 63.0/60.3 63.2/60.5 63.1/60.4 EriCoV NC_022643 Erinaceus europaeus 62.3/57.5 62.2/57.4 62.4/57.2 SC2013 KJ473821 Vespertilio superans 68.2/70.6 68.6/70.8 68.6/71.2 HKU4 NC_009019 Tylonycteris pachypus 96.1/98.4 95.3/97.7 95.5/97.9 HKU5 NC_009020 Pipistrellus abramus 68.9/69.4 69.1/69.3 68.8/69.3 MERS-CoV KJ477102 Camel 67.5/67.2 67.8/67.4 67.5/67.4 MERS-CoV NC_019843 Human 67.4/67.5 67.8/67.6 67.5/67.7 Table 3. Comparison of S gene between HKU4-related strains detected in Macau and other Lineage C betacoronaviruses.
-
Coronaviruses display epithelium tropism and are easily transmitted through oral and fecal excretion. In this study, we detected a high prevalence and diversity of coronavirus infection in bats in Macau. These results expand our knowledge on coronavirus distribution in bats and demonstrate again that bats are natural reservoirs of coronaviruses. In some seasons, bats may carry a high rate of viruses, which may cause a high risk of inter- species transmission if human are highly exposed to bats.
Due to the bat habitat in nature, they usually live close together in large populations, which facilitate virus transmission, both intra- and interspecies. Some bat species may be more susceptible to virus infection and can be infected by more than one virus. In our study, we found a high rate of co-infection by two viruses from two families, coronavirus and adenovirus, in the same individual. It is interesting to determine the mechanism by which these viruses interact in the host.
This preliminary study revealed the prevalence and distribution of viruses in different bat species of Macau. Some of these viruses are more closely related to the viral pathogens of emerging human diseases and may have the capability to cause a cross-species transmission which suggests the potential risk of virus spillover from bats to human population in Macau. From a public health standpoint, long-term surveillance of bat-borne viruses should be performed in the future.
-
We thank Feili Guo, Qiujian Li, Shaofang Hu and Bingrong Liu for helping with the sample collection. We thank Xiangling Liu for helping with viral isolation. We thank Ben Hu for article revision. This work was fi- nanced by the Environment Construction & Capacity Building of GDAS’ Research Platform (2016GDASPT-0215), the Science & Technology Planning Project of Guangdong (2013B050800024 and 2015A020209093) and Science & Technology Planning Project of Guangzhou (201707010128).
-
The authors declare that they have no conflicts of interest. This study was conducted in strict accordance with the ASAB/ABS Guidelines for the treatment of animals in research, and all methods were approved by the Guangdong Entomological Institute Animal Care Committee (Permit number: GDEI-AE-2006001).
-
XLY, LBZ, YW and ZLS designed the experiments. QL, QZ, HL, HPK, KCW, SNC, XYH, XWP, JL and LBZ collected the samples. JL, BL, and XLY performed the experiments, and analyzed the data. JL, ZLS, XLY, LBZ, and YW wrote the paper. All authors read and approved the final manuscript.
Detection of diverse viruses in alimentary specimens of bats in Macau
- Jie Liang 1,2 ,
- Xing-Lou Yang 3 ,
- Bei Li 3 ,
- Qi Liu 2 ,
- Qin Zhang 2 ,
- Hui Liu 2 ,
- Hon-Pio Kan 4 ,
- Kai-Chin Wong 4 ,
- Si-Nga Chek 4 ,
- Xiangyang He 2 ,
- Xingwen Peng 2 ,
- Zheng-Li Shi 3 ,
-
Yi Wu
1,,
 ,
, -
Libiao Zhang
2,,
- Received Date: 12 March 2017
- Accepted Date: 09 May 2017
- Published Date: 01 June 2017
Abstract: Bats carry a variety of viruses,and some of them cause public health problems.Macau,which is famous for its gambling industry,has a complex population structure.The globalization in such an international metropolis has enhanced the chance of disease transmission.Therefore,surveillance of zoonotic viruses is necessary for the early warning of potential emerging infectious diseases. Here,we report the first surveillance of bat viruses in Macau.In this study,we collected 1004 samples involving 10 bat species from 7 sites from April 2015 to May 2016,and examined the presence of viruses using nucleic acid-based methods.Coronaviruses,adenoviruses and paramyxoviruses were detected in these samples,with a high prevalence of coronaviruses.While, none was positive for hepatitis A virus,hepatitis E virus or hantavirus.Co-infections are not common in those bat species,but coronavirus HKU6 and adenovirus can be found commonly occurred in Myotis ricketti.

 DownLoad:
DownLoad: