












-
Dear Editor,
The hepatitis A virus (HAV) is a common agent causing acute liver disease worldwide, with approximately 11, 000 deaths annually (WHO 2017). The virus is transmitted primarily by the fecal-oral route and it normally infects people living in high-density and resource-poor countries (Aggarwal and Goel 2015). Epidemics may also occur after the accidental introduction of HAV to areas with better sanitation but susceptible population (Jacobsen and Wiersma 2010). HAV is a positive-sense, single-stranded RNA virus of the genus Hepatovirus in the family Picornaviridae (Adams et al. 2017). The genome is approximately 7500 nucleotides in length and contains a single open reading frame (ORF) encoding a polyprotein, which is divided into three regions: P1 (VP1–VP4, constituting the viral capsid), P2 (2A–2C), and P3 (3A–3D, constituting the polymerase) (Hollinger and Martin 2013). HAV has been classified as the type species of Hepatovirus (HepV), and historically humans and other primates are the only natural reservoirs for HAV (Hollinger and Martin 2013). Recently, genetically diverse hepatoviruses were reported from bats, rodents, hedgehogs, and shrews worldwide. Eight novel HepV species (HepV B-I) were assigned by the International Committee on Taxonomy of Viruses under the criteria of divergent distance of P1 (> 14%) and P3CD (> 24%) (Adams et al. 2017; Drexler et al. 2015; Yu et al. 2016).
Bats carry a range of important viruses, such as Marburg virus, Hendra virus, and severe acute respiratory syndrome coronavirus (Shi 2013). Although approximately 120 species of bats exist in China, information on bat hepatitis viruses is limited. Recently, two hepatitis agent-related viruses Orthohepadnavirus and Orthohepevirus were identified in Chinese bats (He et al. 2015; Nie et al. 2017; Wang et al. 2017). Here, we report the results of hepatovirus detection in Chinese Hipposideros bats.
In our previous high-throughput sequencing data, we identified a short sequence related to the HAV 3Dpol gene in the feces of Hipposideros armiger in Xianning (unpublished data). Based on this sequence and the published HAV-related sequences, we designed degenerate primers targeting the conserved 3Dpol gene and detected the presence of HAV in fecal samples of Hipposideros bats collected from seven provinces between 2011 and 2014 (Supplementary Table S1). Viral RNA was extracted using the High Pure Viral RNA kit (Roche, Mannheim, Germany). Nested polymerase chain reaction (PCR) was performed using the following primers: HAV-3D-F1 50CYTATHTRAARGATGAGCTKAGA30 and HAV-3DR1 50RTCIAARACWAGRGCNATYG30 (round 1); HAV-3D-F2 50ACRTCATCICCRTARCAIAGRA30 and HAV-3D-R2 50TACCWAATCATRAATGGACT30 (round 2).

Table Table S1 . Hepatovirus detction in Hipposideros bat feces or fecal swab samples
Among the 1266 samples, we detected nine hepatoviruspositive samples (Supplementary Table S1), which shared 99% nucleotide identity with each other and 67%–69% nucleotide identities with other reported hepatoviruses. All the nine sequences were detected in H. armiger feces sampled in Xianning. The sequences were submitted to GenBank under accession numbers MG559666– MG559673. We could not isolate the virus from trials on Vero (African green monkey kidney) and LLC-MK2 (Rhesus monkey kidney) cell lines. The full-length genomic sequence (GenBank accession number: MG559674) named HepV-bat3206 was obtained from a positive sample using a combination of Illumina high-throughput sequencing, genome walking, and 50 rapid amplification of cDNA ends (RACE) (Takara Bio USA, Mountain View, USA).
The full-length genomic sequence of HepV-bat3206 was 7184 nt, which is similar to the size of other reported bat HAV genomes (Drexler et al. 2015). The HepV-bat3206 genome contained 30.44% A, 16.21% C, 22.14% G, and 31.21% U, with a highly biased purine content of 61.65% and a deoptimized codon usage similar to those of other hepatoviruses (Aragones et al. 2010). The genomic organization was identical to those of HepVs, containing a 50-UTR, 30-UTR, and a single ORF encoding the polyprotein layout as 1A–1B–1C–1D/2A–2B–2CHel/3A–3BVPg–3CPro–3DPol. A comparison of the full-length genome of HepVbat3206 with that of hepatoviruses from other organisms showed amino acid and nucleotide similarities in the range of 53%–75% and 58%–70%, respectively (Table 1). The highest amino acid and nucleotide identities were 75.3% and 70.3%, respectively, with HepV-H2_M32Eidhel2010, which was identified in Eidolon helvum from Ghana.

Table 1. Genome and protein similarity between HepVbat3206 and other Hepatovirus species.
To determine the phylogenetic relationship of HepVbat3206 with other hepatoviruses, the polyprotein sequence from representative hepatoviruses, including those from humans, primates, and other small animals, were downloaded from GenBank. Sequence alignment was performed using Clustal X and phylogenetic analysis performed using the default parameters of the MEGA7 software. The phylogenetic tree showed that HepV-bat3206 is distantly related to human and primate hepatoviruses and clustered with bat, Tupia, and hedgehog hepatovirus H (Fig. 1). The phylogenetic tree based on P1 (structural protein) and P3 (3C and 3D, polymerase) also showed a similar topology of the polyprotein tree (Supplementary Figure S1A–S1B). While the P1 and P3 region of HepV-bat3206 shared 83%– 86% and 67%–70% with HepV H1-3, respectively, based on the new species criteria for hepatoviruses (distance of P1 > 14% and P3CD > 24%), HepV-bat3206 was only a new strain of HepV H.

Figure 1. The phylogenetic tree of hepatoviruses based on amino acid sequences of the polyprotein. The neighbor-joining phylogenetic tree was constructed using the MEGA7 with 1000 bootstrap replicates.The virus sequence obtained in this study is indicated by the black solid circle. The TrV was used as an outgroup sequence.
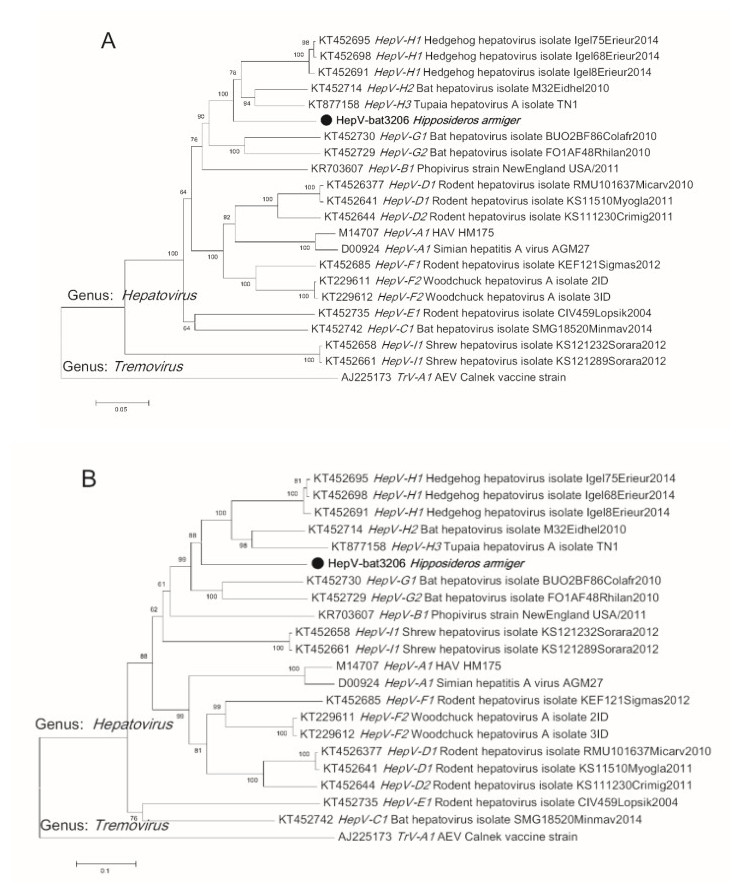
Figure S1. The phylogenetic tree of Hepatovirus based on amino acid sequences of P1 (capsid protein) (A) and P3CD (polymerase protein) (B). The neighbor-joining phylogenetic tree was constructed using the MEGA7 with 1000 bootstrap replicates. The virus sequence obtained in this study is indicated by the black solid circle.
To our knowledge, this is the first report of a bat hepatovirus in China. We detected positive samples in the same colony of bats in the years 2011, 2012 and 2014, suggesting a long history of circulation of hepatoviruses in this colony. Hepatoviruses from nonhuman primates could be transmitted from chimpanzees to humans while the handlers dispose the infected animals (Dienstag et al. 1976). Since the bat cave used for sample collection is a famous tourist site, we suspect a potential risk of transmission of this bat virus to humans if visitors are frequently exposed to this bat colony. Further studies should be focused on the transmission, infection, and prevention of disease caused by these bat hepatoviruses.
HTML
-
This work was jointly funded by the National Natural Science Foundation of China (Grant No. 81290341). We also thank The Core Facility and Technical Support, Wuhan Institute of Virology for assistance in high throughput sequencing sample preparation.
-
The authors declare that they have no conflict of interest.
-
This study was approved by the Animal Ethics Committee of the Wuhan Institute of Virology (Animal ethics Approval Number: WIVA05201202).

 DownLoad:
DownLoad: