Over the past few years, a rising trend of emerging and virulent infectious diseases has been observed. Among the emerging viral infections, more than 75% are natural focal diseases. Natural focal viruses generally recycle in wild animals with relatively large populations as natural hosts, such as bats, rodents, and birds, and are transmitted by vectors such as ticks and mosquitoes that co-exist in the specific natural foci. A program for investigating the main natural hosts and vectors in China has been implemented from 2013 to 2017, which involves a team of researchers across twelve institutions in Xinjiang, Qinghai, Hubei, and Yunnan regions. This special issue collectively presents the interesting results generated from this program. We hope these articles will allow the readers to obtain a better understanding of the importance of investigating pathogen profiles in natural hosts and vectors, and to consider the potential risk of unknown pathogens to the public health. The cover is a gallery of photos which were captured by the team during the field investigation.
Zhiming Yuan. Investigation of Viral Pathogen Profiles in Some Natural Hosts and Vectors in China[J]. Virologica Sinica, 2018, 33(1): 1-4. doi: 10.1007/s12250-018-0021-6.
Over the past few years, a rising trend of emerging and virulent infectious diseases has been observed. Among the emerging viral infections, more than 75% are natural focal diseases. Natural focal viruses generally recycle in wild animals with relatively large populations as natural hosts, such as bats, rodents, and birds, and are transmitted by vectors such as ticks and mosquitoes that co-exist in the specific natural foci. A program for investigating the main natural hosts and vectors in China has been implemented from 2013 to 2017, which involves a team of researchers across twelve institutions in Xinjiang, Qinghai, Hubei, and Yunnan regions. This special issue collectively presents the interesting results generated from this program. We hope these articles will allow the readers to obtain a better understanding of the importance of investigating pathogen profiles in natural hosts and vectors, and to consider the potential risk of unknown pathogens to the public health.
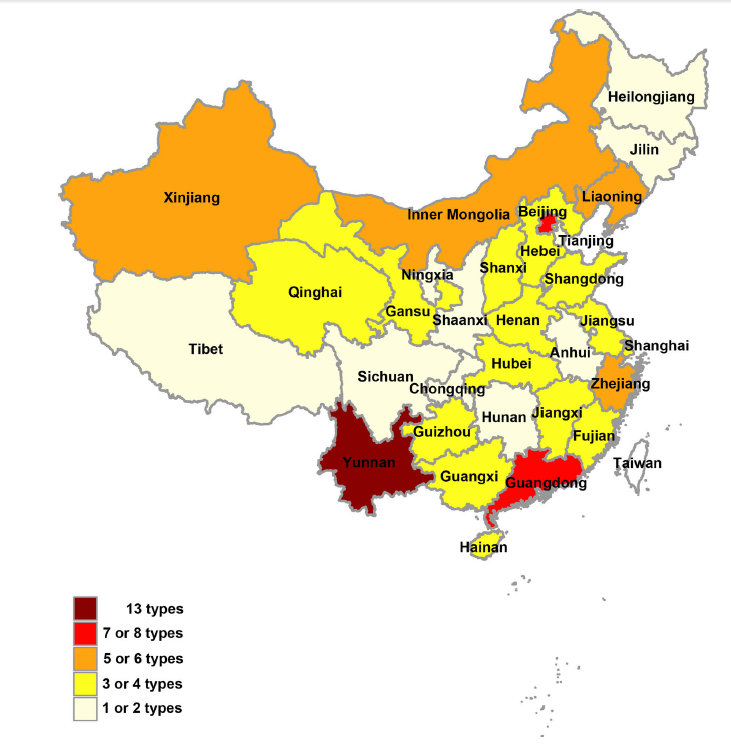
Han Xia, Yujuan Wang, Evans Atoni, Bo Zhang and Zhiming Yuan. Mosquito-Associated Viruses in China[J]. Virologica Sinica, 2018, 33(1): 5-20. doi: 10.1007/s12250-018-0002-9.
Mosquitoes are classified into approximately 3500 species and further grouped into 41 genera. Epidemiologically, they are considered to be among the most important disease vectors in the world and they can harbor a wide variety of viruses. Several mosquito viruses are considered to be of significant medical importance and can cause serious public health issues throughout the world. Such viruses are Japanese encephalitis virus (JEV), dengue virus (DENV), chikungunya virus (CHIKV), and Zika virus (ZIKV). Others are the newly recognized mosquito viruses such as Banna virus (BAV) and Yunnan orbivirus (YNOV) with unclear medical significance. The remaining mosquito viruses are those that naturally infect mosquitoes but do not appear to infect humans or other vertebrates. With the continuous development and improvement of mosquito and mosquito-associated virus surveillance systems in China, many novel mosquito-associated viruses have been discovered in recent years. This review aims to systematically outline the history, characteristics, distribution, and/or current epidemic status of mosquito-associated viruses in China.
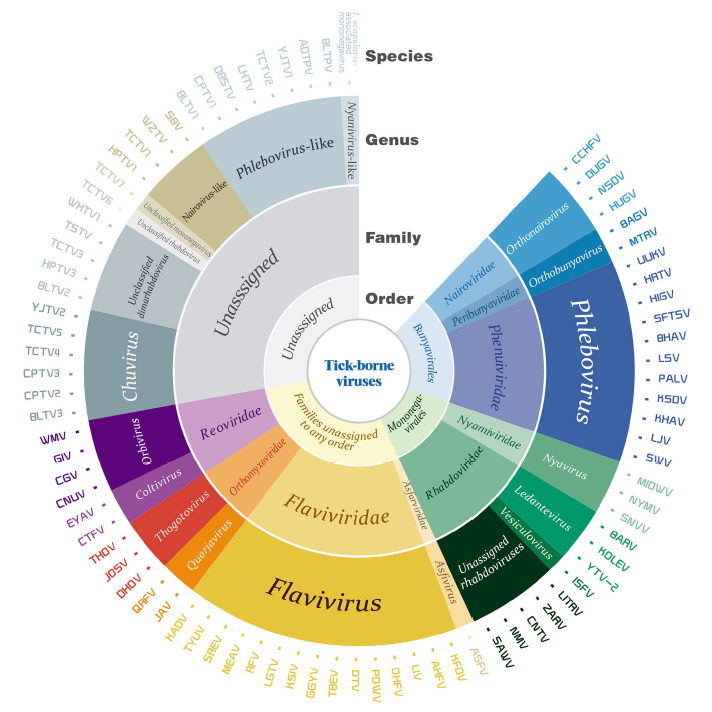
Junming Shi, Zhihong Hu, Fei Deng and Shu Shen. Tick-Borne Viruses[J]. Virologica Sinica, 2018, 33(1): 21-43. doi: 10.1007/s12250-018-0019-0.
Ticks are important vectors for the transmission of pathogens including viruses. The viruses carried by ticks also known as tick-borne viruses (TBVs), contain a large group of viruses with diverse genetic properties and are concluded in two orders, nine families, and at least 12 genera. Some members of the TBVs are notorious agents causing severe diseases with high mortality rates in humans and livestock, while some others may pose risks to public health that are still unclear to us. Herein, we review the current knowledge of TBVs with emphases on the history of virus isolation and identification, tick vectors, and potential pathogenicity to humans and animals, including assigned species as well as the recently discovered and unassigned species. All these will promote our understanding of the diversity of TBVs, and will facilitate the further investigation of TBVs in association with both ticks and vertebrate hosts.
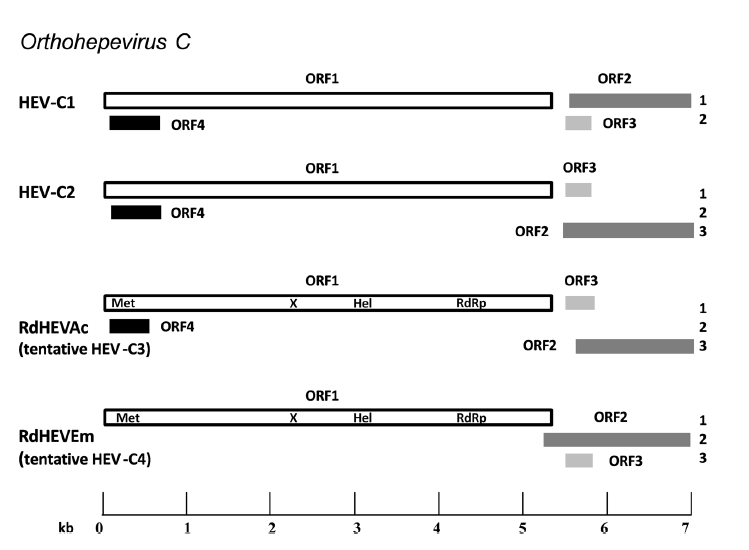
Bo Wang, Wen Li, Ji-Hua Zhou, Bei Li, Wei Zhang, Wei-Hong Yang, Hong Pan, Li-Xia Wang, C. Thomas Bock, Zheng-Li Shi, Yun-Zhi Zhang and Xing-Lou Yang. Chevrier's Field Mouse (Apodemus chevrieri) and Père David's Vole (Eothenomys melanogaster) in China Carry Orthohepeviruses that form Two Putative Novel Genotypes Within the Species Orthohepevirus C[J]. Virologica Sinica, 2018, 33(1): 44-58. doi: 10.1007/s12250-018-0011-8.
Hepatitis E virus (HEV) is the prototype of the family Hepeviridae and the causative agent of common acute viral hepatitis. Genetically diverse HEV-related viruses have been detected in a variety of mammals and some of them may have zoonotic potential. In this study, we tested 278 specimens collected from seven wild small mammal species in Yunnan province, China, for the presence and prevalence of orthohepevirus by broad-spectrum reverse transcription (RT)-PCR. HEV-related sequences were detected in two rodent species, including Chevrier’s field mouse (Apodemus chevrieri, family Muridae) and Père David’s vole (Eothenomys melanogaster, family Cricetidae), with the infection rates of 29.20% (59/202) and 7.27% (4/55), respectively. Further four representative full-length genomes were generated: two each from Chevrier’s field mouse (named RdHEVAc14 and RdHEVAc86) and Père David’s vole (RdHEVEm40 and RdHEVEm67). Phylogenetic analyses and pairwise distance comparisons of whole genome sequences and amino acid sequences of the gene coding regions showed that orthohepeviruses identified in Chinese Chevrier’s field mouse and Père David’s vole belonged to the species Orthohepevirus C but were highly divergent from the two assigned genotypes: HEV-C1 derived from rat and shrew, and HEV-C2 derived from ferret and possibly mink. Quantitative real-time RT-PCR demonstrated that these newly discovered orthohepeviruses had hepatic tropism. In summary, our work discovered two putative novel genotypes orthohepeviruses preliminarily named HEVC3 and HEV-C4 within the species Orthohepevirus C, which expands our understanding of orthohepevirus infection in the order Rodentia and gives new insights into the origin, evolution, and host range of orthohepevirus.
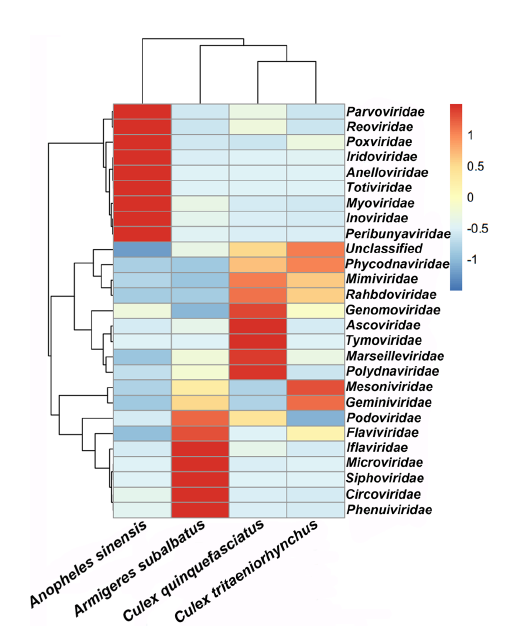
Han Xia, Yujuan Wang, Chenyan Shi, Evans Atoni, Lu Zhao and Zhiming Yuan. Comparative Metagenomic Profiling of Viromes Associated with Four Common Mosquito Species in China[J]. Virologica Sinica, 2018, 33(1): 59-66. doi: 10.1007/s12250-018-0015-4.
Vast viruses are thought to be associated with mosquitoes. Anopheles sinensis, Armigeres subalbatus, Culex quinquefasciatus, and Culex tritaeniorhynchus are very common mosquito species in China, and whether the virome structure in each species is species-specific has not been evaluated. In this study, a total of 2222 mosquitoes were collected from the same geographic location, and RNAs were sequenced using the Illumina Miseq platform. After querying to the Refseq database, a total of 3,435,781, 2,223,509, 5,727,523, and 6,387,867 paired-end reads were classified under viral sequences from An. sinensis, Ar. subalbatus, Cx. quinquefasciatus, and Cx. tritaeniorhynchus, respectively, with the highest prevalence of virus-associated reads being observed in Cx. quinquefasciatus. The metagenomic comparison analysis showed that the virus-related reads were distributed across 26 virus families, together with an unclassified group of viruses. Anelloviridae, Circoviridae, Genomoviridae, Iridoviridae, Mesoniviridae, Microviridae, Myoviridae, Parvoviridae, Phenuiviridae, and Podoviridae were the top ten significantly different viral families among the four species. Further analysis reveals that the virome is species-specific in four mosquito samples, and several viral sequences which maybe belong to novel viruses are discovered for the first time in those mosquitoes. This investigation provides a basis for a comprehensive knowledge on the mosquito virome status in China.
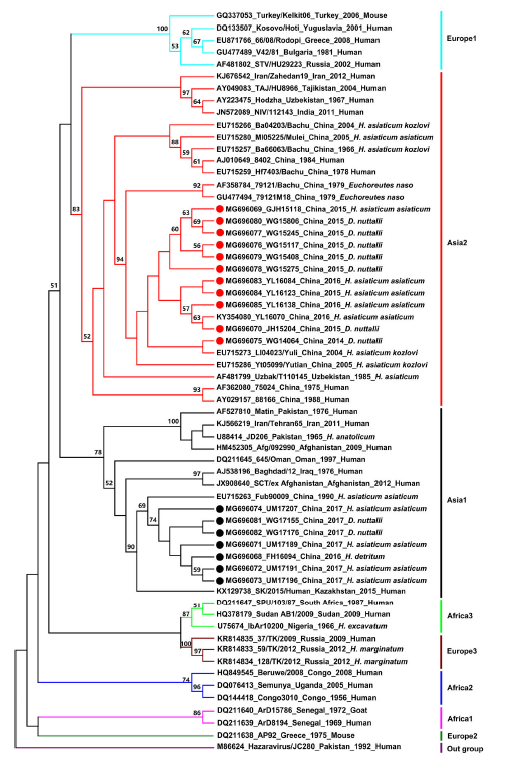
Abulimiti Moming, Xihong Yue, Shu Shen, Chenchen Chang, Cheng Wang, Tao Luo, Yanfang Zhang, Rong Guo, Zhihong Hu, Yujiang Zhang, Fei Deng and Surong Sun. Prevalence and Phylogenetic Analysis of Crimean-Congo Hemorrhagic Fever Virus in Ticks from Different Ecosystems in Xinjiang, China[J]. Virologica Sinica, 2018, 33(1): 67-73. doi: 10.1007/s12250-018-0016-3.
The Crimean-Congo hemorrhagic fever virus (CCHFV), a member of the genus Orthonairovirus and family Nairoviridae, is transmitted by ticks and causes severe hemorrhagic disease in humans. To study the epidemiology of CCHFV in different ecosystems in Xinjiang, China, a total of 58,932 ticks were collected from Tarim Basin, Junggar Basin, Tianshan Mountain, and Altai Mountain from 2014 to 2017. Hyalomma asiaticum asiaticum was the dominant tick species in Tarim and Junggar basins, whereas Dermacentor nuttalli and Hyalomma detritum were found in Tianshan Mountain and Altai Mountain, respectively. Reverse transcription-polymerase chain reaction of the CCHFV small (S) genome segment was used for the molecular detection. The CCHFV-positive percentage was 5.26%, 6.85%, 1.94%, and 5.56% in Tarim Basin, Junggar Basin, Tianshan Mountain, and Altai Mountain, respectively. Sequences of the S segment were used for phylogenetic analysis and the results showed that the newly identified CCHFV strains belonged to two clades. Our study confirms that H. asiaticum asiaticum is the major vector of CCHFV in desert habitats which is consistent with previous studies, and also suggests that H. detritum and D. nuttalli are emerging vectors for CCHFV in Xinjiang. Moreover, this study reports the presence of CCHFV in the mountain habitat of Xinjiang for the first time, suggesting that future surveillance of CCHFV should also include mountainous areas.
Yanfang Zhang, Shu Shen, Yaohui Fang, Jinliang Liu, Zhengyuan Su, Jinhao Liang, Zhong Zhang, Qiaoli Wu, Cheng Wang, Abulikemu Abudurexiti, Zhihong Hu, Yujiang Zhang and Fei Deng. Isolation, Characterization, and Phylogenetic Analysis of Two New Crimean-Congo Hemorrhagic Fever Virus Strains from the Northern Region of Xinjiang Province, China[J]. Virologica Sinica, 2018, 33(1): 74-86. doi: 10.1007/s12250-018-0020-7.
Crimean-Congo hemorrhagic fever (CCHF) caused by the CCHF virus (CCHFV) is a tick-borne natural focal disease with a mortality rate of approximately 50%. CCHFV is widely prevalent in Africa, southern Asia, the Middle East, and southeast Europe. CCHF outbreaks have been reported previously in Xinjiang province, China, especially in its southern region. Epidemiological surveys conducted on ticks and animals have revealed the presence of CCHFV strains in ticks, rodents, and infected individuals from cities and counties in southern Xinjiang. Phylogenetic analyses revealed that the Chinese CCHFV strains belong to one genotype, based on complete sequences of the S segments of its negative-stranded RNA genome. The present study reports two new CCHFV strains isolated from Hyalomma asiaticum asiaticum ticks collected from Fukang City and Wujiaqu City in the northern region of Xinjiang. Viral characteristics and their evolutionary relationships were analyzed through metagenomic and reverse-transcription PCR analyses; these analyses indicated that the genotype of both strains was different from that of other Chinese strains. Furthermore, previous reports of CCHFV in Xinjiang were reviewed and phylogenetic analyses were performed. CCHFV was found to prevail in Fukang City in Junggar Basin for more than 20 years, and that Fukang City and Wujiaqu City are considered natural reservoirs of different genotypes of CCHFV strains. Our findings facilitate the understanding of CCHFV distribution in Xinjiang province and provide insights into the evolutionary relationships among Chinese CCHFV strains.
Yun Luo, Bei Li, Ren-Di Jiang, Bing-Jie Hu, Dong-Sheng Luo, Guang-Jian Zhu, Ben Hu, Hai-Zhou Liu, Yun-Zhi Zhang, Xing-Lou Yang and Zheng-Li Shi. Longitudinal Surveillance of Betacoronaviruses in Fruit Bats in Yunnan Province, China During 2009-C2016[J]. Virologica Sinica, 2018, 33(1): 87-95. doi: 10.1007/s12250-018-0017-2.
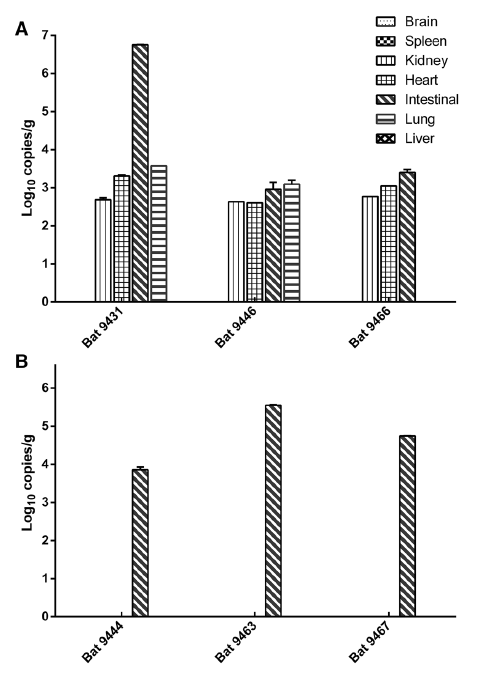
Previous studies indicated that fruit bats carry two betacoronaviruses, BatCoV HKU9 and BatCoV GCCDC1. To investigate the epidemiology and genetic diversity of these coronaviruses, we conducted a longitudinal surveillance in fruit bats in Yunnan province, China during 2009–2016. A total of 59 (10.63%) bat samples were positive for the two betacorona-viruses, 46 (8.29%) for HKU9 and 13 (2.34%) for GCCDC1, or closely related viruses. We identified a novel HKU9 strain, tentatively designated as BatCoV HKU9-2202, by sequencing the full-length genome. The BatCoV HKU9- 2202 shared 83% nucleotide identity with other BatCoV HKU9 stains based on whole genome sequences. The most divergent region is in the spike protein, which only shares 68% amino acid identity with BatCoV HKU9. Quantitative PCR revealed that the intestine was the primary infection organ of BatCoV HKU9 and GCCDC1, but some HKU9 was also detected in the heart, kidney, and lung tissues of bats. This study highlights the importance of virus surveillance in natural reservoirs and emphasizes the need for preparedness against the potential spill-over of these viruses to local residents living near bat caves.
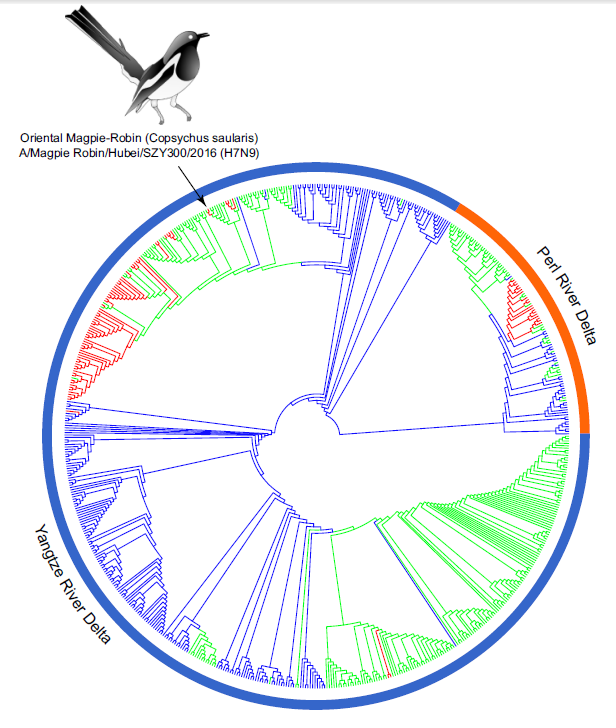
Yanfeng Yao, Tao Zhang, Wenhai Yang, Zhiyong Shao, Bin He, Xiabing Chen, Lijun Wu, Erguang Jin, Haizhou Liu, Jianjun Chen and Jie Chen. Avian Influenza A (H7N9) Virus in a Wild Land Bird in Central China, Late 2015[J]. Virologica Sinica, 2018, 33(1): 96-99. doi: 10.1007/s12250-018-0001-x.
To better understand the role of wild birds in the emergence and potential dissemination of subtype H7N9 viruses, in late 2015, we collected samples from wild land birds in Hubei province and performed virus isolation as well as full genome sequencing. A novel H7N9 virus was isolated from a Magpie-Robin. Genetic analysis showed that the virus was highly similar to the H7N9 viruses that circulated in poultry in other provinces in 2014, suggesting that virus transmission might have occurred between these two regions. The presence of influenza viruses among magpie-robins could increase opportunities for spread to domestic farms and even to humans. This clearly makes it challenging to control the influenza H7N9 subtype. Therefore, it is important to continue monitoring avian influenza virus in wild land birds.
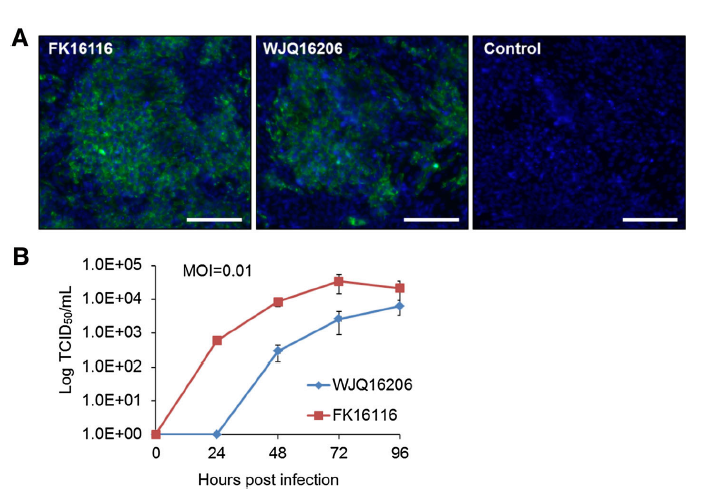
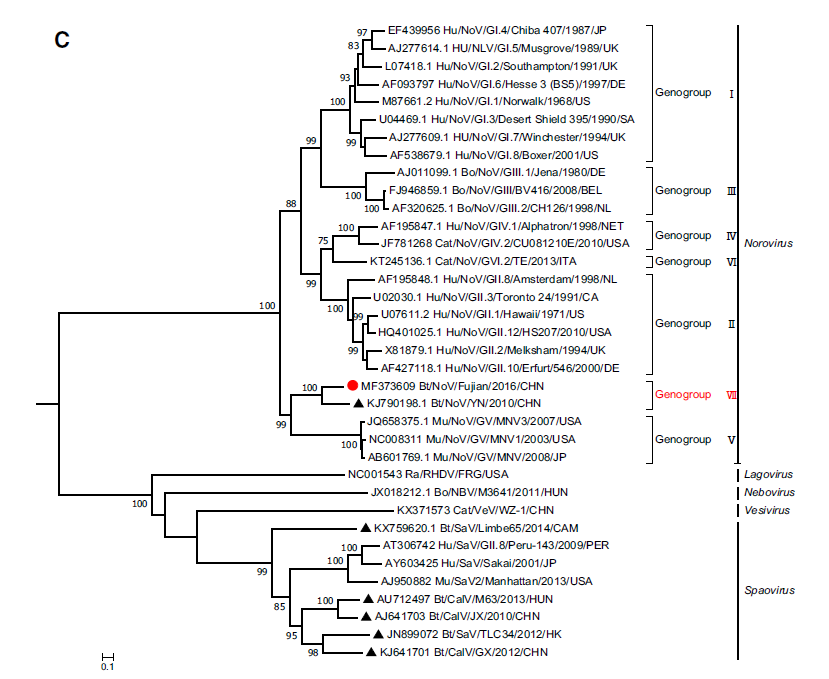
Ling'en Yang, Quanxi Wang, Lin Xu, Changchun Tu, Xiaohong Huang and Biao He. Detection and Characterization of a Novel Norovirus in Bats, China[J]. Virologica Sinica, 2018, 33(1): 100-103. doi: 10.1007/s12250-018-0010-9.
Noroviruses (NoVs) are second only to the rotaviruses as etiologic agents of acute fulminant gastroenteritis in infants and young children worldwide. While only one species is currently recognized within the genus Norovirus, molecular epidemiological studies have demonstrated a marked genetic diversity among circulating NoVs. NoVs have been detected in a number of mammalian species, but only one sequence of bat-borne NoV, from China, has been recorded in Genbank. In this study, we performed NoV screening in 178 adult bats collected from Fujian province, southeast China, and found that 2 bats were positive for this virus. One such sample (NPIH26) was chosen to amplify the whole genome; genomic comparison showed that it possessed a genomic organization typical of noroviruses, and shared the highest nt identity (73%) with the bat NoV BtRs-CalV/YN2010 identified in Rhinolophus sinicus from Yunnan, China. Accordingly, it can be considered as comprising a new genogroup, GVII. This study may help our understanding of evolutionary origins of this virus.
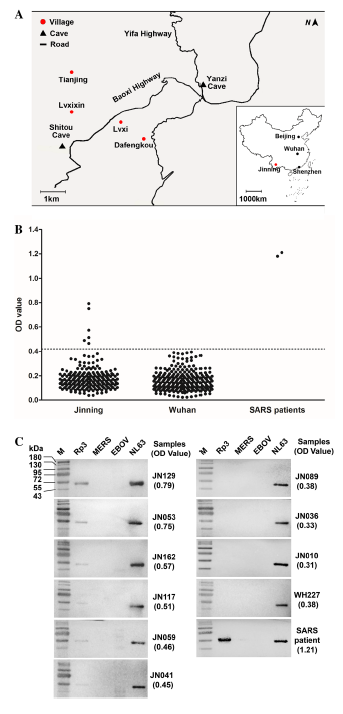
Ning Wang, Shi-Yue Li, Xing-Lou Yang, Hui-Min Huang, Yu-Ji Zhang, Hua Guo, Chu-Ming Luo, Maureen Miller, Guangjian Zhu, Aleksei A. Chmura, Emily Emily, Ji-Hua Zhou, Yun-Zhi Zhang, Lin-Fa Wang, Peter Daszak and Zheng-Li Shi. Serological Evidence of Bat SARS-Related Coronavirus Infection in Humans, China[J]. Virologica Sinica, 2018, 33(1): 104-107. doi: 10.1007/s12250-018-0012-7.
In our previous works, we have reported genetically diverse SARS-related coronaviruses (SARSr-CoV) in a single bat cave, Yunnan province, China, and suggested that some SARSr-CoVs may have high potential to infect humans without the necessity for an intermediate host. In this report, we developed a specific ELISA based on the nucleocapsid protein of a SARSr-CoV strain and detected its antibody in humans who are highly exposed to bat populations. From 218 human serum samples, 6 were positive against the nucleocapsid protein by ELISA and further confirmed by Western blot. For the first time, we demonstrated the SARSr-CoV had spillover to humans, although did not cause clinical diseases.
Wen Li, Bo Wang, Bei Li, Wei Zhang, Yan Zhu, Zheng-Li Shi and Xing-Lou Yang. Genomic Characterization of a Novel Hepatovirus from Great Roundleaf Bats in China[J]. Virologica Sinica, 2018, 33(1): 108-110. doi: 10.1007/s12250-018-0013-6.
The hepatitis A virus (HAV) is a positive-sense, single-stranded RNA virus of the genus Hepatovirus in the family Picornaviridae. HAV is a common agent causing acute liver disease worldwide and primarily transmitted by the fecal-oral route. Approximately 120 species of bats exist in China, but information on hepatovirus in bats is not reported. In this study, we collected 1,266 bat feces or fecal swab samples in eight places of seven provinces in China. Among these 1,266 samples, nine H. armiger feces samples were found positive in Xianning, Hubei. These nine positive samples shared 99% nucleotide identity with each other and 67%–69% nucleotide identities with other reported hepatoviruses. Meanwhile one complete genome of bat hepatovirus named HepV-bat3206 was amplified and analyzed. The complete genome length of HepV-bat3206 is 7,184 nucleotides and shared highest 70% similarity with the reported hepatoviruses. The phylogenetic tree showed that HepV-bat3206 is distantly related to human and primate hepatoviruses and but clustered together with bat, Tupia, and hedgehog hepatovirus H. The positive hepatovirus samples were collected in the same colony of bats in the years 2011, 2012 and 2014, suggesting a long history of circulation of hepatoviruses in this colony. Since the bat cave is a famous tourist site, some measures should be taken to prevent visitors are frequently exposed to the bat feces.
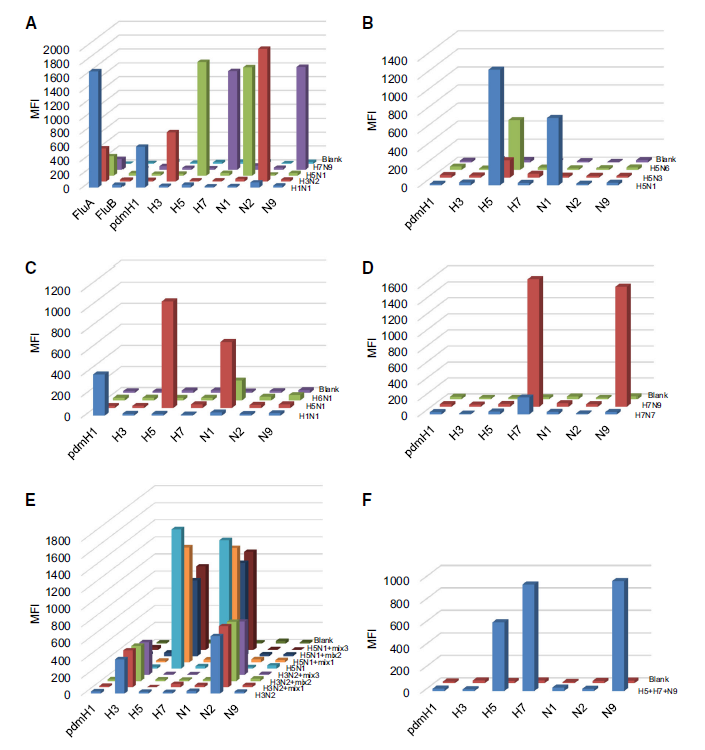
Yi Li, Longquan Ni, Jianjun Chen, Juan Yang, Fei Deng and Hualin Wang. Development of Multi-analyte Suspension Assay for Simultaneously Efficient Detection of Avian Influenza Virus A Subtypes[J]. Virologica Sinica, 2018, 33(1): 111-115. doi: 10.1007/s12250-018-0018-1.
Enormous economic losses and reported cases of bird-to-human transmission alert us the importance of AIVs surveillance in natural foci. However, the limited multiplexing capacity of PCR or real-time PCR method made AIVs surveillance insufficient, costly and labor intensive. In this study, we developed a multi-analyte suspension assay (MASA) to detect AIVs subtypes based on Luminex xMAP technology. With this MASA, 7 HA and 5 NA subtypes could be identified in a single reaction. The evaluation data showed that these subtype-specific amplified primers had high specificity and the coverage of each primer pairs ranged from 82% to 97%. Our studies provide a rapid and economical method for AIVs surveillance of natural foci.