












HTML
-
Crimean-Congo hemorrhagic fever is a severe hemorrhagic disease caused by Crimean-Congo hemorrhagic fever virus (CCHFV) infection, with a fatality rate of 5%–30% (Ergonul et al. 2006; Paragas et al. 2004). CCHFV is widely distributed in more than 30 countries in Africa, Asia, the Middle East, and Eastern Europe (Lani et al. 2015; Messina et al. 2015; Calisher and Goodpasture 1975). In China, CCHFV is also known as the Xinjiang hemorrhagic fever virus (XHFV), as it was first discovered in Xinjiang, China in 1965 (Bao et al. 2011). Subsequent morphological and serological studies showed that XHFV was identical with CCHFV (Yu et al. 2006). Previous studies have shown that the prevalence of CCHF, a typical tick-borne natural focaldisease, is closely associated with tick activity, and the distribution characteristics of ticks varied in different ecosystems (Sun et al. 2009).
With complex landforms and various habitats, the Xinjiang area breeds abundant natural hosts and vectors of viruses and other pathogenic microorganisms (Dai et al. 2006). Previous investigations showed that the Tarim and Junggar basins are natural desert foci of CCHFV, with Hyalomma asiaticum asiaticum as the main vector (Sun et al. 2009). However, studies on the diversity of tick species in different habitats in the area, ranging from basin arid desert to alpine forest steppe, are limited.
In this study, molecular epidemiology of CCHFV from ticks collected in Tarim Basin, Junggar Basin, Tianshan Mountain, and Altai Mountain from 2014 to 2017 was analyzed. The results showed that H. asiaticum asiaticum was the dominant tick in Tarim and Junggar basins, while Dermacentor nuttalli and H. detritum were dominant in the Tianshan Mountain and Altai Mountain, respectively. CCHFV prevalence in different ticks in various habitats was comprehensively evaluated and a phylogenic analysis was performed. The results indicated the presence of two main CCHFV genotypes circulating in Xinjiang, carried by different ticks in different ecosystems.
-
Xinjiang province is located in the northwest border of China. This study focused on four geographic areas (the arid desert in Tarim Basin and Junggar Basin, mountain forest steppe of Tianshan Mountain, and hilly arid desert steppe of Altai Mountain) that cover 17 counties or cities (Fig. 1). Based on the geographical distribution of ticks in Xinjiang (Yu et al. 1997), 16, 041, 12, 343, 13, 850, and 16, 698 ticks were collected between April and May of 2014, 2015, 2016, and 2017, respectively. Ticks were collected using the artificial flag method according to their morphological characteristics, collection time and place, and quantity of ticks, and were stored at -80 ℃.
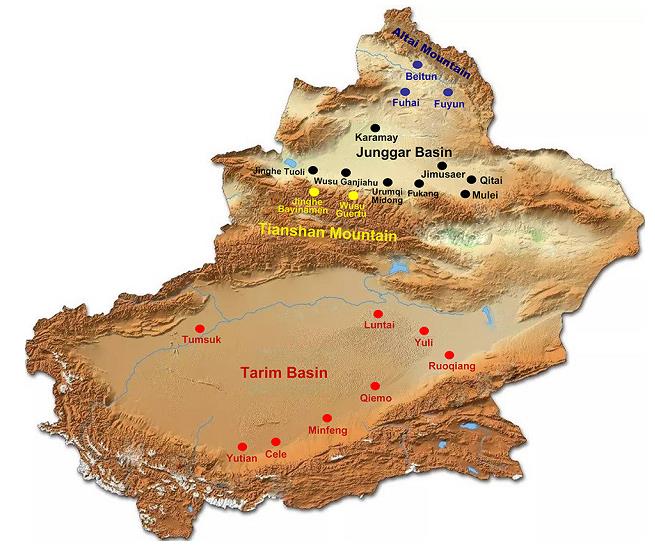
Figure 1. Map of the Xinjiang province of China showing sampling locations for the current study. Red dots indicate sampling spots in the arid desert of Tarim Basin, black dots indicate sampling spots in the arid desert of Junggar Basin, yellow dots indicate sampling spots in the north slope forest grassland of the Tianshan Mountain, and blue dots indicate sampling spots in the drought desert grassland in hilly south slope of the Altai Mountain. The county names are shown.
-
Generally, 50–100 ticks grouped as a pool were put into a 5-mL grinding tube containing 2 mL Dulbecco modified Eagle medium (DMEM; Gibco, Carlsbad, CA, USA). The homogenates of each pool were prepared using a tissue grinder (D1000; NZK, China) and centrifuged at 4 ℃, 3300×g for 10 min. The supernatants were collected and stored separately. The Trizol reagent (Invitrogen, Carlsbad, CA, USA) was used for RNA extraction as follows: 500 μL Trizol and 200 μL centrifuged tick suspension supernatant were added into an RNase-free microcentrifuge tube (Thermo Fisher Scientific, Waltham, MA, USA), followed by addition of 400 μL chloroform and mixing with manual agitation for eight times. The tube was incubated at 20 ℃ for 3–5 min, followed by centrifugation at 13, 200×g for 15 min at 4 ℃. The supernatant was transferred into a new microcentrifuge tube, and 1 mL isopropanol was added and incubated at -80 ℃ for 1 h or at -20 ℃ overnight. After centrifugation at 13, 200×g for 10 min at 4 ℃, l mL 75% ethanol was added to the extract, followed by centrifugation at 13, 200×g for 5 min; the supernatant was discarded. After nucleic acid precipitation at 55 ℃ for 10 min, 33 μL diethyl pyrocarbonate (DEPC)-treated water was added to dissolve the RNA for reverse transcription. cDNA was synthesized using M-MLV reverse transcriptase (TaKaRa Biotechnology, Dalian, China) according to the manufacturer's instructions. Briefly, 3 μL dNTP mixture, 10 μL 5× reverse transcriptase buffer, 2 μL random primer (50 mmol/L), 1 μL MLV reverse transcriptase, 1 μL RNAase inhibitor, 20 μL total RNA, and 13 μL RNAasefree water were mixed and sequentially incubated at 37 ℃ for 50 min and then at 72 ℃ for 10 min. The cDNA was subsequently stored at -20 ℃.
-
All tick pools were analyzed for the presence of CCHFV RNA. A 220-bp region of CCHFV small (S) segment was amplified by specific nested PCR. PCR was performed in a 50-μL reaction mix containing 1–3 μL cDNA template from each pooled sample, 25 μL of 2 × mix buffer, and 20 pmol each of the forward and reverse primers (Tsingke Biotechnology Co., Ltd. Wuhan, China). For the first round of PCR, a primer pair (CCHFV-F2 5'-TGGACACCTTCA CAAACTC-3' and CCHFV-R3 5'-GACAAATTCCCTG CACCA-3') (Ma et al. 2001) was used to amplify a 530-bp product. The PCR amplifications were performed in a thermocycler (Bio-Red, Hercules, CA, USA) using the following program: denaturation at 95 ℃ for 5 min, followed by 30 cycles of denaturation at 95 ℃ for 30 s, annealing for 30 s, and extension at 72 ℃ for 30 s, with a final extension at 72 ℃ for 8 min. For the second round of PCR, the nested-primer pair (CCHF-F3-nest 5'-GAA TGTGCATGGGTTAGCTC-3' and CCHF-R2-nest 5'-GA CATCACAATTTCACCAGG-3') (Guo et al. 2017) was used to amplify a 220-bp fragment. The PCR amplifications were performed using the following program: denaturation at 95 ℃ for 5 min, followed by 30 cycles of denaturation at 95 ℃ for 30 s, annealing for 30 s, and extension at 72 ℃ for 20 s, with a final extension at 72 ℃ for 5 min. The PCR products were analyzed by electrophoresis using a 1.0% agarose gel stained with ethidium bromide.
-
The amplified products were sequenced by Sanger sequencing (Tsingke Biotechnology Co., Ltd. Wuhan, China), and sequences were compared with those found in the GenBank database by nucleotide sequence homology searches performed at the network server of the National Center for Biotechnology Information (NCBI) using BLAST analysis (http://www.ncbi.nlmn.nih.gov/BLAST). The sequences were edited and aligned using the BioEdit software (Hall 1999). A phylogenetic analysis of CCFHV was conducted using the sequence data obtained in this study and representative CCHFV S gene sequences from GenBank, and the NCBI nucleotide BLAST program. ClustalW was used for generating the S partial sequence alignment, and a scaled phylogenetic tree of the CCHFV strains based on the 220-bp S RNA sequences (corresponding to nucleotides 329 to 548 of CCHFV Chinese strain 7001) was generated using the maximum likelihood (ML) method with Kimura 2-parameter distance on the MEGA6 software (Tamura et al. 2013). Bootstrap confidence limits were based on 1000 replicates. This method evaluated the topologies of different trees and selected the best tree based on a specified model according to the evolutionary process that can account for the interconversion of sequences (Sadegh et al. 2016).
-
The correlation between CCHFV-positive rates by RTPCR in ticks from different sampling sites was estimated using the Pearson product-moment correlation coefficient, and the independent Chi square test was used to determine the significance of the correlation (Sun et al. 2009).
Investigation Areas and Sampling
Sample Processing and RNA Extraction
Polymerase Chain Reaction (PCR)
Sequencing and Phylogenetic Analyses
Statistical Analyses
-
From 2014 to 2017, a total of 58, 932 tick samples were collected from 17 counties in four geographic ecosystems in Xinjiang, including the arid desert area in Junggar Basin and Tarim Basin, the north slope forest grassland of the Tianshan Mountain, and drought desert grassland in hilly south slope of the Altai Mountain. Among the collected ticks, 10, 840 samples of H. asiaticum asiaticum were grouped into 130 pools, 47, 608 D. nuttalli samples were grouped into 464 pools, and 454 H. detritum samples were grouped into 18 pools, and all pools were screened for the presence of CCHFV RNA by nested RT-PCR (Guo et al. 2017). The distribution of CCHFV in four geographical ecosystems was assessed; H. asiaticum asiaticum was mainly distributed in the arid desert region in Tarim and Junggar basins, whereas D. nuttalli and H. detritum were mainly distributed in the north slope forest grassland of the Tianshan Mountain and the drought desert grassland in the hilly south slope of the Altai Mountain. The CCHFVpositive percentage was 5.26%, 6.85%, 1.94%, and 5.56% in Tarim Basin, Junggar Basin, Tianshan Mountain, and Altai Mountain, respectively. The results are shown in Table 1.

Table 1. Molecular detection of CCHFV from ticks isolated from different regions in Xinjiang, China.
-
The tick populations in the abovementioned ecosystems were different. H. asiaticum asiaticum was the predominant tick species in the arid desert region in Tarim and Junggar basins, whereas D. nuttalli and H. detritum were dominant in the north slope forest grassland of the Tianshan Mountain and the drought desert grassland in the hilly south slope of the Altai Mountain (Table 1). CCHFV was mainly detected in H. asiaticum asiaticum, D. nuttalli, and H. detritum, and the CCHFV-positive samples included 8 of 130 (6.15%) H. asiaticum asiaticum pools, 9 of 464 (1.94%) D. nuttalli pools, and 1 of 18 (5.56%) H. detritum pools. The results showed that the positive detection rate of CCHFV in H. asiaticum asiaticum and H. detritum was significantly higher (χ2 = 6.948, P = 0.031) than that in D. nuttalli (Table 1), suggesting that the Hyalomma tick is the major CCHFV vector in Xinjiang. Nevertheless, this study was the first to detect CCHFV in H. detritum and D. nuttalli in the Altai Mountain and Tianshan Mountain.
-
The PCR product obtained using the CCHFV-S primers was recovered and sequenced. Eighteen partial S segments obtained from this study were analyzed. The results indicated that the amplified fragment (220 bp) from 18 Chinese strains had high similarity in terms of nucleic acid (85%– 99.5% identity) and amino acid sequences (88.9%–100% identity).
Sequences of CCHFV S segment obtained in this study were submitted to GenBank and used for a phylogenetic analysis (Supplementary Table S1). Figure 2 shows the phylogenetic tree based on the 18 partial S segments from this study and 44 representative CCHFV sequences from GenBank (Supplementary Table S2). Among the 18 partial sequences, nine were from D. nuttali, eight from H. asiaticum asiaticum, and one from H. detritum (Supplementary Table S1).
Our results showed that CCHFV strains could be generally grouped into eight clades: Europe 1, Asia 2, Asia 1, Africa 3, Europe 3, Africa 2, Africa 1, and Europe 2 (Fig. 2). This result is in agreement with that of previous studies (Hewson et al. 2004; Guo et al. 2017; OrKun et al. 2017). Based on the result of the sequencing and phylogenetic analyses, CCHFV strains obtained in this study were clustered into two clades. Eleven newly detected Chinese strains from Wusu Ganjiahu, Wusu Guertu, and Jinghe Bayinamen in the Junggar Basin, and from Yuli in the Tarim Basin were clustered in the Asia 2 clade, which contains strains from Xinjiang, China, as well as from Uzbekistan, Tajikistan, India, and other countries. Other novel Chinese strains from Wusu Guertu and Urumqi Midong in the Junggar Basin, and Fuhai in the Altai prefecture were clustered in the Asia 1 clade, which also contains strains from Xinjiang, China, as well as from Pakistan, Oman, Kazakhstan, and others. In both clades, sequences from different tick species appeared to be grouped together, indicating that emergence of the two CCHFV clades may be independent of their vectors.
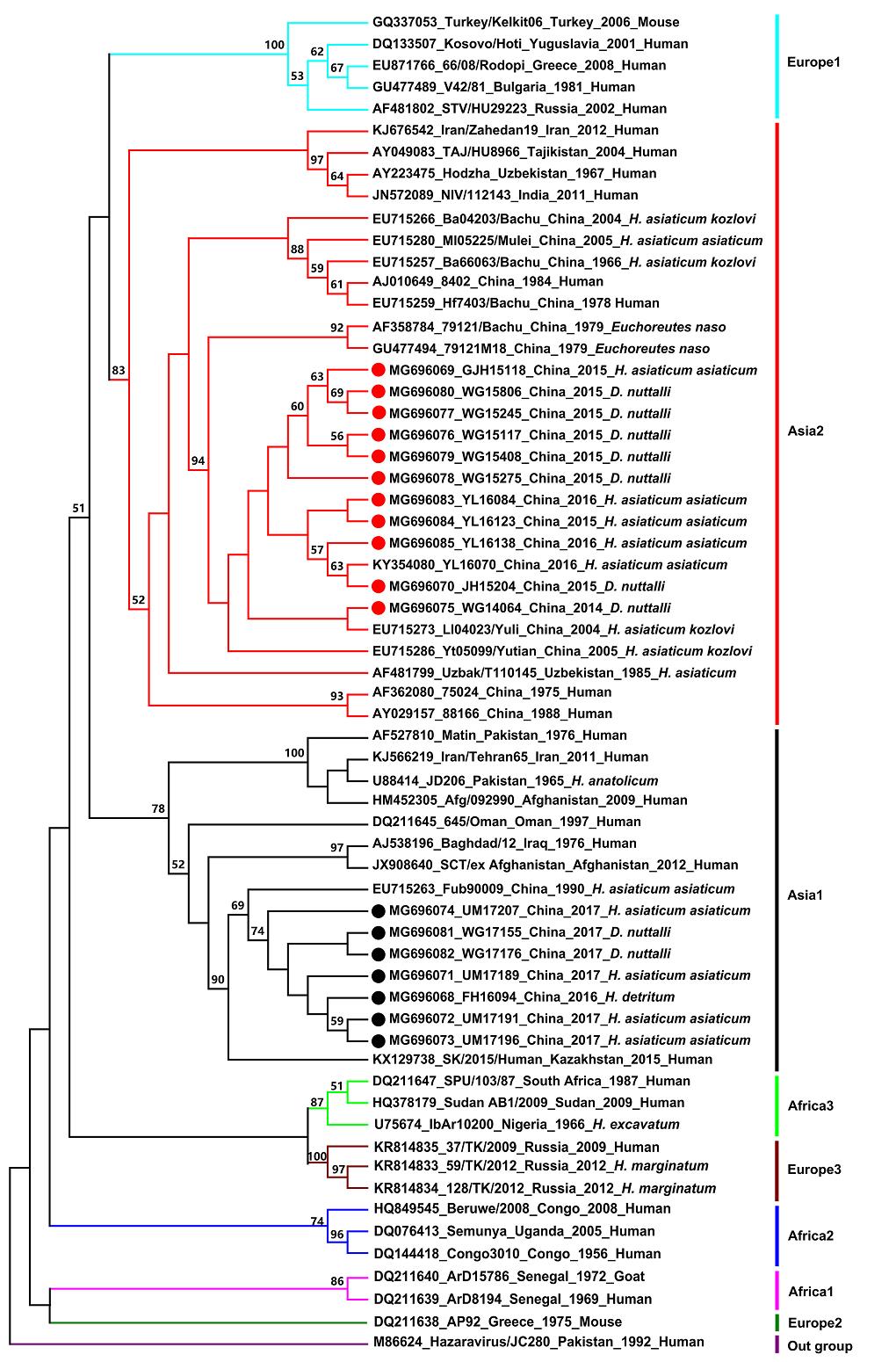
Figure 2. Phylogenetic tree of CCHFV strains based on sequences of the 220-nucleotide S RNA fragment. Strains are displayed in the format of "GenBank accession number, strain, country, year, and origin". The tree was constructed using the maximum likelihood method with MEGA6 software. Values lower than 50% are not shown. The red and black circles indicate sequences obtained in this study (detailed information in Supplementary Table S1), while others are from reference strains (detailed information in Supplementary Table S2).
Geographical Distribution of CCHFV
Distribution of CCHFV Host Ticks
Phylogenetic Tree Mapping Based on CCHFV S Fragment Sequences
-
CCHF caused by CCHFV is a naturally-occurring epidemic disease that is distributed globally. CCHFV can be carried and spread by more than 30 different tick species in various ecological environments (Hoogstraal 1979; Turell et al. 2007; Sun et al. 2011). The Xinjiang region in China has diverse natural and ecological landscapes that include alpine basins, desert regions, and wetlands, which are favorable for different tick species to survive. Previous studies demonstrated that more than 40 species of ticks are important natural vectors and reservoirs for various zoonotic pathogens in Xinjiang (Yu et al. 1997); nevertheless, despite having a significant epidemic potential and being tick-borne, human cases of CCHF were only reported in the Tarim Basin of Xinjiang. Therefore, whether geographic distribution of CCHF is associated with different tick species in different eco-geographical landscapes in Xinjiang remained unclear.
Previous studies showed that H. asiaticum asiaticum was widely distributed in Xinjiang (Kou et al. 2016; Yu et al. 2006). Furthermore, CCHFV has been isolated from a large number of H. asiaticum asiaticum collected in the Tarim Basin, indicating that H. asiaticum asiaticum is the major vector and host of CCHFV in this area of Xinjiang (Guo et al. 2017; Sun et al. 2006). The geoecological landscape of the Junggar Basin in northern Xinjiang is comparable to the arid desert landscape of the Tarim Basin. In 1990, Feng et al. (1991) isolated a new CCHFV strain from H. asiaticum asiaticum collected in the Fukang County in Junggar Basin. In 2009, Sun et al. (2009) conducted an epidemiological investigation of CCHFV in the Junggar Basin and found that the positive detection rate of CCHFV in H. asiaticum asiaticum was 5.6% by nested PCR, and the detection rate in sheep by serological testing was 3.6%, demonstrating that CCHF is also common in the Junggar Basin. The present study showed that the positive detection rate of CCHFV in H. asiaticum asiaticum in the Junggar Basin was 6.85%. Furthermore, two CCHFV strains were isolated from H. asiaticum asiaticum collected in Fukang and Wujiaqu counties in the Junggar Basin (unpublished data). These results confirmed that Junggar Basin was also a natural focus of CCHFV, and similar to that observed in the Tarim Basin, H. asiaticum asiaticum was the main host and vector of CCHFV in the Junggar Basin.
The geoecological landscape of the Tianshan Mountain in the southern margin of Junggar Basin gradually evolves into mountain forest grassland, where D. nuttalli is the dominant tick species. In this study, the CCHFV detection rate in D. nuttalli was 1.94%, which was significantly lower than that in H. asiaticum asiaticum (P = 0.031). In contrast, the geoecological landscape of the Altai Mountain in northern Junggar Basin gradually evolves into arid hilly grassland, with H. detritum as the predominant tick species. In the present study, we found that the detection rate of CCHFV in H. detritum was 5.56%, which was not significantly different from that in H. asiaticum asiaticum (P > 0.05).
In this study, CCHFV was detected for the first time in D. nuttalli and H. detritum, indicating that these ticks were also vectors for CCHFV in Xinjiang. With environmental changes affecting both vectors and host, CCHFV has demonstrated extensive adaptability during the course of evolution; this is in line with the diverse ecologicalgeographic landscape of CCHFV distribution and characteristics of the vector and host diversity (Sun et al. 2011). However, since no cases of CCHFV human infection were reported in these areas, the role of these tick species on CCHFV epidemic in Xinjiang requires further investigation.
In this study, sequences of 18 CCHFV strains from different geographical regions (Tarim Basin, Junggar Basin, Tianshan Mountain, and Altai Mountain) were determined, and the genetic diversity and phylogenetic relationships among 62 CCHFV strains from different countries (obtained in this study and from GenBank) were analyzed (Fig. 2). The sequence analysis indicated that the 18 Chinese strains shared high nucleotide (85%–99.5%) and amino acid identities (88.9%–100%). Although the three tick species evaluated in this study are found in four different habitats that are geographically distant and environmentally diverse, the phylogenetic tree analysis showed that the 18 CCHFV strains were clustered into two clades (Asia 1 and 2). Notably, 11 Xinjiang strains grouped together with CCHFV strains from Uzbekistan, Tajikistan, and India, while seven other Xinjiang strains clustered with strains from Pakistan, Oman, and Kazakhstan, indicating that CCHFV circulating in Xinjiang and neighboring countries are genetically related. These results are similar to those obtained in an earlier study (Sun et al. 2009).
In conclusion, we identified the prevalence of CCHFV in different dominant ticks from four different geographic ecosystems and their genetic characteristics. Our results indicate a natural prevalence of CCHFV in the study sites, and confirm for the first time the presence of CCHFV epidemic in a mountain habitat area (Tianshan Mountain and Altai Mountain) in Xinjiang. Based on the results of previous surveys, we speculate that the main maintenance host and vector of CCHFV in Xinjiang include H. asiaticum asiaticum, H. detritum, and D. nuttalli. However, there are few comprehensive studies on the molecular or serological detection of CCHFV in rodents, domestic animals, and humans in Xinjiang, which requires further investigation and additional data collection in future CCHFV studies. Thus, further epidemiological investigations of tick diversity and animal reservoir of CCHFV in various habitats are needed to better understand the genetic diversity of CCHFV.
-
This work was supported by the Ministry of Science and Technology of China (2013FY113500), the grants from the National Science Foundation of China (Nos. 81460303, 81760365), and Funded by the Open Research Fund Program of the State Key Laboratory of Virology of China (No. 2015IOV003).
-
SRS, FD, YJZ and ZHH designed the experiments. AM and CCC carried out the experiments. YJZ, XHY, TL and CW collected the samples. RG, SS and YFZ provided experimental and sequence analysis support. SRS and YJZ analyzed the data. AM and SRS wrote the paper. All authors read and approved the final manuscript for submission.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of interest
Animal and Human Rights Statement
-

Table S1. Xinjiang CCHFV strains detected in the present study

Table S2. CCHFV isolates from GenBank used for the phylogenetic analysis

 DownLoad:
DownLoad: