












HTML
-
Mosquitoes, which can be classified into approximately 3500 species and further grouped into 41 genera, are the most common and important vectors for viruses. Mosquitoes carry or transmit a wide variety of viruses which belong to two categories. The first category includes the viruses that can replicate in both mosquito and vertebrate cells, or can infect humans or other vertebrates. Most of the viruses in this group are known as pathogens of humans or animals, such as yellow fever virus (YFV), dengue virus (DENV), West Nile virus (WNV), chikungunya virus (CHIKV), Zika virus (ZIKV), Rift Valley virus (RVFV), Akabane orthobunyavirus (AKAV), Tembusu virus (TMUV), and Banna virus (BAV), within the families Flaviviridae, Togaviridae, Reoviridae, and Phenuiviridae (Conway et al. 2014). The second category comprises of the insect-specific viruses (ISVs), which only naturally infect mosquitoes, and replicate in mosquito cells in vitro, but cannot replicate in vertebrate cells, or infect humans or other vertebrates. They are widely distributed in the families of Birnaviridae, Flaviviridae, Mesoniviridae, Parvoviridae, Peribunyaviridae, Reoviridae, Rhabdoviridae, Togaviridae, and Tymoviridae (Bolling et al. 2015).
An. sinensis mainly inhabits rice fields, ditches, streams, irrigation canals, marshes, ponds, and ground pools. They are a zoophilic mosquito which prefer to bite warm-blooded animals such as cattle (Zhu et al. 2013). An. sinensis is the main vector for malaria, but is also considered to be a vector for viruses such as Japanese encephalitis virus (JEV), BAV, and Kadipiro virus (KDV) (Liu et al. 2013; Zhang 1990). Ar. subalbatus commonly live in the places close to human dwellings, especially in sub-urban areas with poor sanitation and polluted water sites. They prefer to feed on humans or livestock (Rajavel 1992). Ar. subalbatus is known to be the vector of JEV and Getah virus (GETV) (Chen et al. 2000; Zheng et al. 2015). Cx. quinquefasciatus is widely distributed globally in populated regions and is often closely associated with bites on humans, other mammals, or birds (Farajollahi et al. 2011). C. quinquefasciatus is known to be an important vector of WNV. It also transmits St. Louis encephalitis virus, western equine encephalitis virus, and ZIKV (Samy et al. 2016). Cx. tritaeniorhynchus inhabits wells, ponds, and urban environments close to human settlements. It is known to be the main vector of JEV, and also a potential vector for BAV and TMUV (Liu et al. 2010; Self et al. 1973; Tang et al. 2013). All four mosquito species are extremely common and widespread in China.
Recently, the rapid development and application of next generation sequencing (NGS) technology has enabled broad surveys of viral diversity and has significantly increased our knowledge of viruses present in vectors, such as mosquitoes and ticks (Brinkmann et al. 2016; Xia et al. 2015). For example, detection of the presence of vertebrate viruses, originating from the blood of mosquitoes' host, which then accumulate in the mosquitoes' intestine during blood feeding, is possible (Chandler et al. 2015; Ng et al. 2011; Shi et al. 2015). In addition, recently, multiple unclassified mosquito-associated virus sequences, such as Wuhan mosquito virus, Xinzhou mosquito virus, Zhejiang mosquito virus, Zhee Mosquito virus, Wutai Mosquito virus, and Cx. tritaeniorhynchus rhabdovirus, have been found (Shi et al. 2016). In our previous study, a metagenomic survey of mosquitoes collected in different cities in the Hubei province, the results indicated a high abundance and diversity of viruses in mosquitoes, and several novel viruses or sequences were identified (Shi et al. 2015). However, the characteristic features of the virome associated with different mosquito species in same geographic locations has not been well investigated and evaluated.
In this study, a metagenomic comparison analysis for the virome structure associated with four different mosquito species (An. sinensis, Ar. subalbatus, Cx. quinquefasciatus, and Cx. tritaeniorhynchus) collected in Yichang region was carried out, and the results indicated distinct virome profiles in each species, indicating that the differences among viromes are species-specific.
-
A total of 2222 adult mosquitoes were collected from sites adjacent to human settlements, cowsheds, and pigpens, in Yichang region of Hubei Province in China. Sampling was done in the months of June to September 2014. No protected species were sampled. The mosquito capture method involved the use of electrical mosquito aspirators (Yalin, China). The collected mosquitoes were transported to laboratory alive inside net traps, then euthanized by freezing at -20 ℃ for 30 min. They were then morphologically classified and further grouped into pools of approximately 50 adult mosquitoes per pool. The mosquito pools were subsequently washed once in 70% ethanol and distilled water, for 5 min each, to remove environmental contaminants.
-
The pools of mosquitoes in each species were homogenized and centrifuged as described previously (Shi et al. 2015). After centrifugation, the supernatant was filtered using a 0.22-μm polycarbonate filter membrane (Millipore, Billerica, USA) to remove bacteria and other debris present. RNA was extracted from 140 μL of supernatant. The QIAamp viral RNA extraction kit (Qiagen, Hilden, Germany) was used for RNA extraction according to the manufacturer's instructions. The final RNA elution was done with 60 μL AVE buffer. The RNA samples were sent to the Shanghai Biotechnology Corporation for library construction and sequencing by paired-end mode through the Illumina Miseq platform (Illumina, San Diego, USA).
-
Sequencing adaptors were trimmed and reads with low quality or match to the mosquito genome were discarded, to acquire clean reads for each sample. De novo assembly was then used to obtain contigs. These steps were conducted by Shanghai Biotechnology Corporation.
The Kaiju web server was used for taxonomic classification of the reads and contigs, and the Greedy mode was chosen with SEG filter, minimum match length = 11, minimum match score = 75, and allowed mismatches = 5 (Menzel et al. 2016). The Kaiju RefSeq Genome (protein sequences from 7065 complete bacterial and archaeal genomes, and 9334 viral genomes from NCBI RefSeq, updated 2017-05-16) was selected as the reference database (http://kaiju.binf.ku.dk/). Blastx (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to make a double confirmation for the contigs matching to viral sequences. The extraction for the information of assigned reads, which classified them to a viral family and species, together with other statistical analyses, were conducted in R, version 3.2.5 (https://www.r-project.org/). Metagenomic comparative analysis was conducted through MetaComp (http://cqb.pku.edu.cn/ZhuLab/MetaComp/) under the multisample statistic method (Zhai et al. 2017).
Mosquito Sampling and Taxonomy Identification
Nucleic Acid Extraction and Sequencing
Data Analysis
-
A total of 2222 mosquitoes were collected and identified to species level through morphological identification under a stereomicroscope. They were classified into four species: An. sinensis, Ar. subalbatus, Cx. quinquefasciatus, and Cx. tritaeniorhynchus, with 387, 852, 745, and 238 mosquitoes from each species, respectively. The four species are very common in the Yichang region, Hubei, and the dominant species in 2014 were Ar. subalbatus (38.3%) followed by Cx. quinquefasciatus (33.5%).
-
A total of 6, 535, 325, 7, 183, 603, 6, 714, 707 and 8, 909, 955 clean, paired reads and 251, 198, 284, and 186 assembled contigs were generated from An. sinensis, Ar. subalbatus, Cx. quinquefasciatus, and Cx. tritaeniorhynchus, respectively, through Illumina Miseq sequencing. After querying of the RefSeq Complete Genomes database through the Kaiju Web Server, the result indicated that there were many reads that corresponded to known viral sequences (Table 1). The percentage of the reads that matched to viral sequences were 52.5%, 30.9%, 85.3%, and 71.1% in An. sinensis, Ar. subalbatus, Cx. quinquefasciatus, and Cx. tritaeniorhynchus respectively, and highest prevalence of virus associated reads were observed in Cx. quinquefasciatus.

Table 1. Reads/contigs in each mosquito species.
-
Taxonomic classification was conducted based on viral families. The results indicate that the virus-related reads present in the four mosquito viromes are distributed across 26 virus families, together with an unclassified group (Supplementary Table S1).
Anelloviridae, Circoviridae, Genomoviridae, Iridoviridae, Mesoniviridae, Microviridae, Myoviridae, Parvoviridae, Phenuiviridae, and Podoviridae were the top ten significantly different viral families (Fig. 1). According to the detailed analysis results in Supplementary Table S1, many reads related to Iridoviridae, Poxviridae, Totiviridae, and Peribunyaviridae were found in An. sinensis, while they were rarely detected in the other three species. In addition, the reads that matched to Phenuiviridae and Siphoviridae were identified in Ar. subalbatus, however rarely found in the other species.
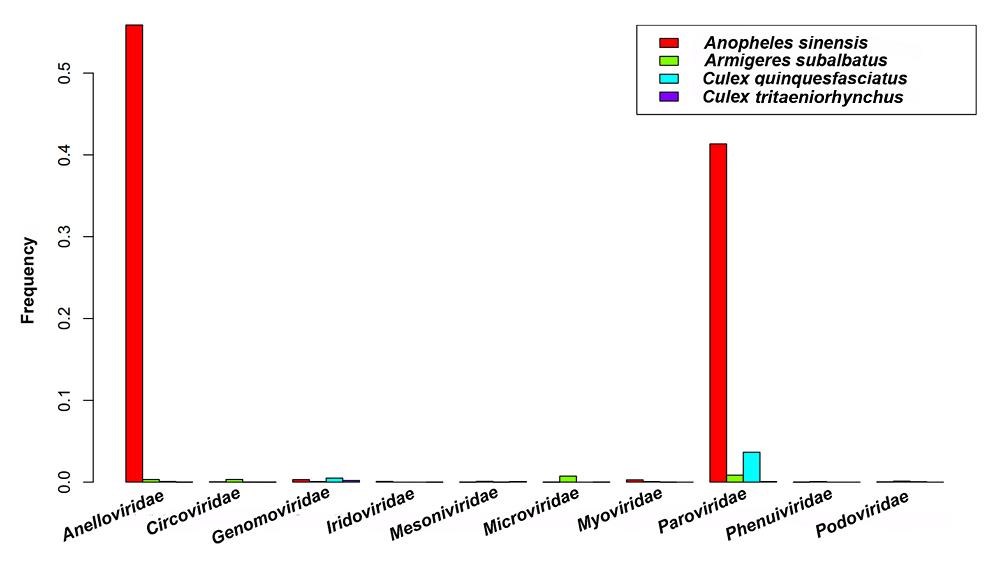
Figure 1. Top ten significantly different viral families among four mosquito species. Frequency is the rate of the number of reads belong to a viral family to the total number of viral reads in one mosquito sample. The frequencies of Anelloviridae and Parvoviridae in four samples dramatically fluctuated.
To detect differences in virome structures among the different mosquito species, a taxonomic heatmap and hierarchical cluster analysis were conducted based on a distance matrix (Fig. 2). The heatmap analysis based on virus family showed that each species had a special virome structure. For instance, in An. sinensis mosquitoes, Parvoviridae, Reoviridae, Poxviridae, Iridoviridae, Anelloviridae, Totiviridae, Myoviridae, Inoviridae, and Peribunyaviridae were found to be present in abundant quantities while in Ar. subalbatus, Iflaviridae, Microviridae, Siphoviridae, Circoviridae, and Phenuiviridae were the most abundant virus families. However, the abundant viral families in Cx. quinquefasciatus species were found to be Genomoviridae, Ascoviridae, Tymoviridae, Marseilleviridae, and Polydnaviridae. In Cx. tritaeriorhynchus mosquitoes, Mesoniviridae and Geminiviridae were detected in abundant quantities. The cluster results showed Cx. quinquefasciatus and Cx. tritaeniorhynchus grouped together, which indicated the correlation of the virome structures in these two species were closer than they were to the other species, to some extent.For example, the Phycodnaviridae, Mimiviridae and Rhabdoviridae viral families were found to be in abundant or moderate levels in both Culex species.
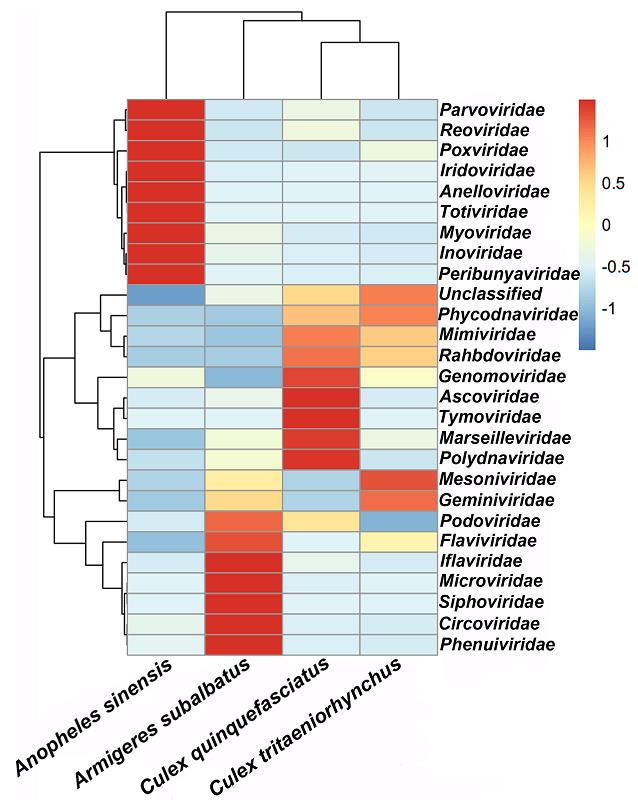
Figure 2. Heatmap based on 26 viral families. Heatmap was constructed based on the distance matrix, which was calculated by Euclidean distance method according to the number of reads belonging to each viral family in four mosquito species. The X axis represents the sample names of mosquito species, the Y axis represents the names of viral families. The color red to blue stands for the highest to lowest abundant of viral reads in each species according to viral family.
-
The information for contigs that matched to the known viruses in each sample is summarized in Table 2. In An. sinensis, three types of Torque teno sus virus (1a, 1b, and k2) were detected, however, none of them were detected in Cx. tritaeniorhynchus. Three types of mesonivirus were found in Cx. tritaeniorhynchus, and two types in Ar. subalbatus, but none of them were detected in An. sinensis or Cx. quinquefasciatus. The Wuhan fly virus was only detected in An. sinensis. BAV was found in An. sinensis and Cx. tritaeniorhynchus. Large amount of contigs matched to Cx. tritaeniorhynchus rhabdovirus, Wenzhou sobemo-like virus 3, and Wuhan mosquito virus 8 in the two Culex species. Hubei mosquito virus 2 was found in all four species. Many newly identified viruses, such as the Hubei mosquito virus, Hubei tombus-like virus, Wenzhou sobemo-like, and Zhejiang mosquito virus reported by Shi et al. (2016), were detected in our mosquito samples (Table 2).

Table 2. Information on contig hits to the virus database in four mosquito species.
In addition, several viral sequences which maybe belong to novel viruses (low identity to known virus) for the first time presented in mosquitoes were discovered (Table 2). For example, the Tanay virus was only previously reported in Culex spp. or Armigeres spp. in the Philippines (Li et al. 2014). However, there was one contig matched with Tanay virus (with 46% identity) in our An. sinensis sample. Several contigs hit to Wuhan fly virus 1, Wuhan ant virus, Beihai mantis shrimp virus 6, Hubei picorna-like virus 41, Hubei rhabdo-like virus 1, Sanxia water strider virus 8, or Wuhan house centipede virus 6, with the identity ranging from 29% to 76%, which was the first time that these viruses have been found in mosquitoes. In future work, we will try to isolate them and conduct further analysis.
Taxonomic Identification of Mosquitoes
Illumina Sequencing Results and Viral-Related Reads/Contigs in Four Mosquito Species
Comparative Analysis for the Viral Family Profile Based on Reads
The Viral Species Profile in Four Mosquito Species Based on Assembled Contigs
-
Mosquitoes are known to obtain blood meal from vertebrates, which in turn are known to be natural reservoirs of the majority of viruses that are transmitted by invertebrate vectors. Hence, the diversity for mosquito virome could be influenced by the communities of vertebrates present. Most of the reported metagenomic analyses of the mosquito virome compared the diversity among the mosquitoes from different locations or ecological systems (Chandler et al. 2015; Frey et al. 2016; Ng et al. 2011; Shi et al. 2015), in which it is difficult to infer whether the differences between viromes were caused by the ecological communities in different locations or by the mosquito species. Here, we describe the biodiversity of the viral communities associated with An. sinensis, Ar. subalbatus, Cx. quinquefasciatus, and Cx. tritaeniorhynchus mosquitoes, which inhabit Yichang and commonly attack people or livestock in this region. We detected the virome differences among the four species and the results indicated that each species had a specific taxonomic structure, and that they contained sequences related to a broad range of animal, plant, insect, bacterial, fungal, and protozoan viruses. In addition, the clustering algorithms demonstrated that the distance between the viromes of Cx. quinquefasciatus, and Cx. tritaeniorhynchus, were much closer than between the other species, which may be because of them being in the same genus.
BAV is the member of genus Seadornavirus in Family Reoviridae, and its genome has 12 dsRNA segments.Itwasfirst isolated from the cerebrospinal fluid and serum of encephalitis patients in Xishuangbanna, Yunnan, in 1987. It is considered to be a new pathogen, causing human viral encephalitis and fever (Liu et al. 2010; Xu et al. 1990). Subsequently, BAV has been isolated from 10 mosquito species in 3 genera (Cx. tritaeniorhynchus, Cx. pipiens pallens, Cx. annulus, Cx. pseudovishnui, Cx. modestus, An. sinensis, Ae. vagus, Ae. albopictus, Ae. vexans, and Ae. Dorsalis) mosquitoes from China (Liu et al. 2010). In this study, the virus sequences associated to BAV within the family Reoviridae were detected in all four mosquito species tested, and the highest abundance was found in the Ar. subalbatus mosquito species (Supplementary Table S1 and Table 2), which provides further evidence for the widespread nature of BAV in different mosquito species. In addition, this result indicated the potential risks of BAV infection in the local residents of Yichang and that serological study should be conducted in the future.
Mesoniviridae is a newly classified viral family. The members in this family consist of ssRNA of ~ 20 kb genome size, and they replicate in mosquitoes. Mesoniviruses have been proposed to provide an evolutionary link between nidovirus lineages, with small and large RNA genome sizes (Lauber et al. 2012; Nga et al. 2011). A novel species of mesonivirus named Yichang virus was isolated from Culex spp. from the sample which was the same batch used for the NGS analysis here (Wang et al. 2017). The metagenomic study showed the Mesoniviridae related sequences were present in all four mosquito species, and they had a much higher prevalence in Ar. subalbatus and Cx. tritaeniorhynchus, with identity ranging from 50% to 67% (Supplementary Table S1 and Table 2), which indicated that the mesonivirus is commonly distributed in mosquitoes in this region.
Viral sequences belonging to unclassified virus were detected in all four species, but the prevalence of unclassified virus was much higher in the sample from Ar. subalbatus, Cx. quinquefasciatus and Cx. tritaeniorhynchus when compared to An. sinensis. Most of these sequences are closely related to the newly identified virus by Shi et al. (2016), such as the Wuhan mosquito virus, Hubei mosquito virus, Zhejiang mosquito virus, and Wenzhou sobemo-like virus. This indicated that these types of viruses are extremely common in mosquitoes, and further work for towards their isolation and characterization should be done to investigate the role of their presence in mosquito.
In our previous study, the presence of vertebrate viruses, such as Anelloviridae and Parvoviridae originating from the blood of the mosquitoes' host, were found in the mosquito virome. A large number of viral sequences, which were closely related to the torque teno sus virus (distributed in pigs) or porcine parvovirus, were found in the sample from Anopheles spp. This might be because of pigs being one of the main hosts for the blood feeding of Anopheles mosquito, and that the infection rate for these two types of viruses in pigs is high in this region.
In summary, we sequenced and analyzed the mosquito virome from four species, and we found that the virome structure varied greatly among mosquito species (An. sinensis, Ar. subalbatus, Cx. quinquefasciatus, and Cx. tritaeniorhynchus) in the same geographic regions. This approach could help us to gain a better understanding of the virus flora in mosquito species.
-
We thank the Hubei Provincial Center for Disease Control and Prevention for assistance in mosquito sampling within the Hubei Province. This work was supported by the Ministry of Science and Technology of China (Science and Technology Basic Work Program 2013FY113500).
-
HX and ZY designed the experiments. YW, and LZ, carried out the experiments. HX, YW, CS, and AE analyzed the data. HX, YW and ZY wrote the paper. HX and CS checked and finalized the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.


 DownLoad:
DownLoad: