












HTML
-
Ticks (Parasitiformes order, Ixodidae family) act as parasites as well as vectors of pathogens for humans, domestic animals, and wildlife, such as Borrelia duttoni, Babesia, African swine fever virus, Crimean–Congo hemorrhagic fever virus, severe fever with thrombocytopenia syndrome virus, and Nairobi sheep disease virus (Dantas-Torres et al. 2012; Luo et al. 2015; Shi et al. 2018). However, in addition to their role as transmission vectors, ticks harbor a large number of viromes, including those from many insect-specific viruses (Ailawadi et al. 2015; Pettersson et al. 2017). The worldwide documentation of tick-borne disease is increasing, as a result of the discovery of new tick-associated medium and increased incidences of tickborne diseases (Kilpatrick and Randolph 2012). In the United States, ticks transmit the vast majority of arthropodborne pathogens and are associated with the most frequently occurring tick-borne illness (Eisen et al. 2017). In South Asia, the Haemaphysalis spinigera tick transmits Kyasanur Forest disease virus, which causes severe hemorrhagic fever with neurological manifestations (Shah et al. 2018). Although these insect-specific viruses have no direct impact on public health, they may regulate the spread of the viruses that cause disease in vertebrates (Tree et al. 2016). Therefore, in order to prevent tick-borne infectious diseases, it is crucial to compare the viromes in ticks and the tick vectors.
The development of metagenomic sequencing approaches has been widely used and has served as a tool for reevaluating the tick virome, resulting in the novel discoveries of viruses in the unclassified Chuvirus and Negevirus groups (Zhang et al. 2018). Viral metagenomics has been used to detect viral pathogens, unknown pathogenic viruses, and viruses of unknown cellular origins (Rosario and Breitbart 2011; Bibby and Peccia 2013). Through tickvirome analysis, it was previously found that ticks harbor a large number of highly diverse viruses, including novel and highly divergent viruses that infect vertebrate and invertebrate hosts (Tokarz et al. 2014), animal insect and plant virus (Xia et al. 2015). Recent analysis of tick viromes conducted in Heilongjiang Province, China revealed novel viruses and several likely novel tick-borne viruses, such as Beiji phlebovirus and Jingmen tick virus (Meng et al. 2019). In addition, using viral metagenomic analysis, Tan et al. identified a new strain of Wenzhou virus harbored by rodents in Xinjiang, China (Tan et al. 2019). However, studies of tick viruses in central and eastern China are limited.
Thus, to gain insight into the natural ecology of tick viruses, we employed a viral metagenomic approach to characterize the whole virus environment within tick samples. We characterized the total viromes in five tick species collected from four locations in central and eastern China. Specifically, we determined the number, type, and abundance of each virus within tick samples and addressed whether these parameters varied by species and sampling location. Our assessment highlights advances in molecular pathogen detection that can be made using viral metagenomic approaches, which may lead to the discovery of several new tick-borne disease agents.
-
We collected 573 adult ticks in 2017 from four locations in central and eastern China that are considered to be of significant public health risk in relation to tick-borne diseases, including Lyme disease, tick-borne encephalitis, and East Coast fever (Table 1). The four locations comprised (ⅰ) Shanghai (location A), an important financial center in eastern China with a large population; (ⅱ) Hebei (location B), an eastern-coastal region with recently increasing cases of tick-borne death; (ⅲ) Henan (location C), an critical economic province in central China with multiple cases of tick-borne death since 2010; and (ⅳ) Hubei (location D), a central location with strong industry in terms of animal husbandry.
Pool name Number of ticks Host Area Location Tick01 93 Domestic dogs Shanghai A Tick02 94 Domestic dogs Hebei B Tick03 95 Domestic sheep Hebei B Tick04 96 Domestic sheep Henan C Tick05 97 Domestic cattle Hubei D Tick06 98 Domestic dogs Henan C Table 1. Sampling information of ticks.
Live ticks were sampled from domestic animals at tickinfested locations by corralling a herd and "scratching" each animal individually to detect attached ticks. Ticks were placed in labeled vials and left on dry ice until they were returned to the laboratory, where the samples were placed in a - 80 ℃ freezer. Tick species identification was initially carried out using taxonomic keys and dissecting microscopes on clod tables and was later verified by sequencing the cytochrome c oxidase subunit I (COI) gene, which encodes a subunit of the cox1 protein. Five species of ticks were represented by the ticks sampled from the indicated geographic locations in central and eastern China (Fig. 1).
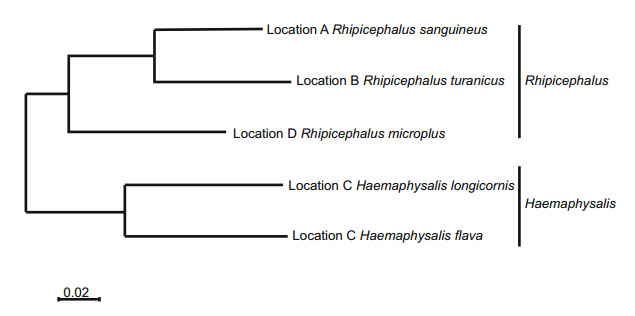
Figure 1. Maximum-likelihood phylogenetic map for the cytochrome c oxidase (COX 1) gene from the tick samples collected in this study. The sampling location and tick species are indicated in each case.
-
As shown in Table 1, these ticks sampled from different domestic animals comprised six pools: Shanghai domestic dogs (n = 93; pool Tick01), Hebei domestic dogs (n = 94; pool Tick02), Hebei domestic sheep (n = 95; pool Tick03), Henan domestic sheep (n = 96; pool Tick04), Hubei domestic cattle (n = 97; pool Tick05), and Henan domestic dogs (n = 98; pool Tick06). Sequencing were performed with 10 to 20 representative ticks from each pool. Prior to homogenization, each tick pool was washed with 75% alcohol, followed by three washed with 1 mL phosphatebuffered saline to remove external microbes. The tick samples were homogenized, frozen, and thawed three times on dry ice, and the supernatants were then collected after centrifugation (10 min, 15, 000 ×g). All library preparations were conducted in the Shanghai Veterinary Research Institute, Shanghai Academy of Agricultural Sciences.
-
Five hundred microliters of each supernatant were filtered through a 0.45-lm filter (Millipore) to remove eukaryotic and bacterial cell-sized particles. The filtrates enriched in viral particles were treated with DNase and RNase to digest unprotected nucleic acid at 37 ℃ for 100 min (Deng et al. 2015). The remaining total nucleic acid was isolated using the QIAamp Mini Viral RNA Kit (Qiagen), according to the manufacturer's protocol. Six libraries were then constructed using the Nextera XT DNA Sample Preparation Kit (Illumina) and sequenced using the MiSeq Illumina platform with 250-base pair (bp), paired-end reads and dual barcoding for each pool. For bioinformatics analysis, the bar codes were removed from the 250-bp, paired-end reads using software from Illumina. An in-house analysis pipeline running on a 32-node Linux cluster was used to process the data. Reads were considered duplicates if bases 5 to 55 were identical and only one random copy of duplicates was kept. The cleaned reads within each barcode were assembled de novo using the ENSEMBLE assembler (Finn et al. 2011). Contigs and all singlet reads were matched against a customized viral proteome database using BLASTx with an E value cutoff of < 10-5, where the virus BLASTx database was compiled using a NCBI virus reference proteome (ftp://ncbi.nih.gov/refseq/release/viral/) to which was added viral protein sequences from an NCBI nr fasta file (based on annotation taxonomy in the virus kingdom). Candidate viral hits were then compared to an in-house non-virus non-redundant (NVNR) protein database to remove false-positive viral hits, where the NVNR database was compiled using non-viral protein sequences extracted from an NCBI nr fasta file (based on annotation taxonomy, excluding the virus kingdom). Contigs without significant BLASTx similarity to the viral proteome database were searched against viral protein families in the vFam database using HMMER3 to detect remote viral protein similarities. The genome coverage of the target viruses was analyzed using Geneious software (Biomatters).
-
Standard precautions were used for all procedures to prevent the cross-sample contamination and nucleic acid degradation. Mainly, serosol filter pipet tips were used to reduce the possibility of sample cross contamination, and all the materials (including the microcentrifuge tubes and, pipet tips) which that directly contacted with the nucleic acid samples were RNase- and DNase- free.
-
Phylogenetic analyses were performed in this study, based on the predicted amino acid or nucleotide sequences. The closest viral relatives were determined based on BLASTx searches in GenBank, and representative members of related viral species or genera. Sequence alignment was performed using CLUSTAL W, with the default settings. Phylogenetic trees with 500 bootstrap replicates of the alignment data sets were generated using the maximum-likelihood method in MEGA7.0. Bootstrap values (based on 1000 replicates) are shown for each node.
-
Putative ORFs in the viral genome were predicted using Geneious 8.1 software or NCBI ORF finder. Putative exon and intron were predicted with Netgenes2 at http://www.cbs.dtu.dk/services/NetGene2/. The conserved domains of bole tick virus 3 from the ticks were determined using the NCBI conserved-domain search in combination with the Pfam conserved-domain search (Finn et al. 2014). Mapping raw data to the reference virus genome was performed using the low sensitivity/fastest parameter in Geneious software version 8.1.
-
Genome sequences for ticks collected in locations A–D were deposited in NCBI GenBank under accession numbers SRR9669407, SRR9669408, SRR9669409, and SRR9669406, respectively.
-
Raw sequence reads from the metagenomic libraries were deposited in raw libraries (https://viralmetagenomics.net/viral/171113_Wen_mixedsamples/Tick01_blast_filter.html, https://viralmetagenomics.net/viral/171113_Wen_mixedsamples/Tick02_blast_filter.html, https://viralmetagenomics.net/viral/171113_Wen_mixedsamples/Tick03_blast_filter.html, https://viralmetagenomics.net/viral/171113_Wen_mixedsamples/Tick04_blast_filter.html, https://viralmetagenomics.net/viral/171113_Wen_mixedsamples/Tick05_blast_filter.html, and https://viralmetagenomics.net/viral/171113_Wen_mixedsamples/Tick06_blast_filter.html). These data are open source and can be downloaded from NCBI.
Tick Collection
Tick Sample Pool Preparation
Viral Metagenomic Analysis
Quality Control in for the Nucleic Acid Manipulation
Phylogenetic Analysis
Other Sequence Analysis
Accession Numbers
Availability of Data and Materials
-
A total of 13, 721 reads from the ticks in all six pools were obtained through Illumina MiSeq sequencing of the nucleic acid libraries from the tick samples. Subsequent BLAST analyses revealed the viromes of 19 virus families, including 2319 sequencing reads annotated as vertebrate viruses, 11, 176 sequencing reads annotated as insect viruses, 72 sequencing reads annotated as plant viruses, and 62 sequencing reads annotated as phages (Fig. 2). Additionally, 92 reads were annotated as algae and fungi (data not shows). Subsequent BLAST analyses revealed that complete L genes of two new species of RNA viruses, both of which are described here. One new virus species fell into the Chuviridae family, whereas the other fell into the Phenuiviridae family. In each pool, the abundance (i.e., frequency) of each virus varied from 0 to 91.4% of all viral reads within the pool (Table 2). This finding suggests that the marked variation in viral abundance was likely related to the sampling location. The virus species detected in this study fell into a wide range of RNA virus groups, including known families and orders, namely, Chuviridae, Mononegavirales, Ortervirales, Picornavirales, Caudovirales, Herpesvirales, retro-transcribing viruses, and other positive-sense RNA viruses.

Figure 2. The composition and distribution of viruses detected in each tick pool. The pie chart in the center showed the approximate percentages of the four virus groups detected in all six pools. The four circumjacent pie charts show the approximate percentages of the indicated types of virus sequences from different pools. The data for each pool are represented with different colors.
Classification Rhipicephalus Haemaphysalis Locations (Host) R. sanguineus
Location A (Dog)R. turanicus
Location B (Dog)R. turanicus
Location B (Sheep)R. microplus
Location D (Cattle)H. longicornis
C (Sheep)H. flava
Location C (Dog)NSRV1 Chuviridae 91.4 73.66 87.26 1.98 0.64 0 NSRV2 Phenuiviridae 1.55 13.88 4.33 0 0 0.35 All viruses 92.95 87.54 91.59 0 0 0 The percentage of total reads were shown.
NSRV negative-sense RNA virus.Table 2. Presence and abundance of newly discovered RNA viruses in different tick species from domestic animals.
For each library, the number of viral families varied from 8 to 19 (Table 3). The abundance (i.e., number) of sequencing reads for each viral family also varied from 1 to 1783. In comparison, the number of sequencing reads that did not correspond to a known viral family varied from 1 to 6590, which often contained the new species not covered by existing genera, suggesting the importance of identifying potentially novel viruses in the viral reservoirs. Indeed, for individual viral families, the abundance levels were comparable across libraries, including both highly abundant viruses such as Phenuiviridae (1 to 1783) and those of lower abundance such as Microviridae (1 to 2). Overall, for all libraries from the four different locations, the total abundance levels of viral RNA sequences were above 37.74%, suggesting that RNA viruses could make up a substantial part of the RNA environments in the ticks.
Family Classification Reads numbers for different viral families Location A (Dog) Location B (Dog) Location B (Sheep) Location C (Sheep) Location D (Dog) Location D (Cattle) None DNA virus 0 0 2 3 0 6 RNA virus 1531 6590 2721 1 2 76 Unclassified virus 13 89 42 4 3 14 Phenuiviridae ssRNA virusa 50 1783 202 2 1 1 Rhabdoviridae ssRNA virus 7 67 20 0 1 16 Retroviridae ssRNA virus 4 13 2 42 22 16 Picornaviridae ssRNA virus 0 0 0 11 10 6 Flaviviridae ssRNA virus 0 0 0 0 0 4 Paramyxoviridae ssRNA virus 0 0 0 1 0 0 Poxviridae dsDNA virus 0 1 0 9 6 30 Iridoviridae dsDNA virus 5 1 0 13 10 8 Phycodnaviridae dsDNA virus 0 1 0 4 6 23 Caulimoviridae dsDNA virus 1 1 1 15 8 4 Siphoviridae dsDNA virus 0 0 0 1 0 26 Myoviridae dsDNA virus 0 0 0 0 0 26 Papillomaviridae dsDNA virus 0 5 7 13 0 0 Polydnaviridae dsDNA virus 0 0 0 0 7 5 Alloherpesviridae dsDNA virus 0 0 0 3 4 2 Herpesviridae dsDNA virus 0 0 0 3 4 1 Microviridae ssDNA virus 2 0 1 1 0 1 Parvoviridae ssDNA virus 2 0 1 0 0 0 All viruses 1615 8551 2999 126 84 263 DNA deoxyribonucleic acid, dsDNA double-stranded deoxyribonucleic acid, ssRNA single-stranded ribonucleic acid. Table 3. Presence and abundances of viral families from different locations.
In addition, some of the viruses were highly prevalent. In particular, Phenuiviridae/Phlebovirus, Retroviridae, and Caulimoviridae were found among all pools, whereas Rhabdoviridae, Baculoviridae, and Iridoviridae were present in five out of six pools. Importantly, negative-sense RNA virus 1 (NSRV1) was detected in most pools and represented the majority of reads in the three pools from Shanghai and Hebei (1478, 6304, and 2617, respectively). This observation highlights the persistence of some novel viral infections in the Shanghai ticks and Hebei ticks.
-
Generally, ticks from dogs contained more viruses than ticks from cattle and sheep (Table 3, Fig. 3A). Of the 19 viral families, only 11, Phenuiviridae, Rhabdoviridae, Retroviridae, Baculoviridae, Poxviridae, Iridoviridae, Phycodnaviridae, Caulimoviridae, Alloherpesviridae, Herpesviridae, and Microviridae were shared between the three hosts. However, the abundances of these viromes varied.
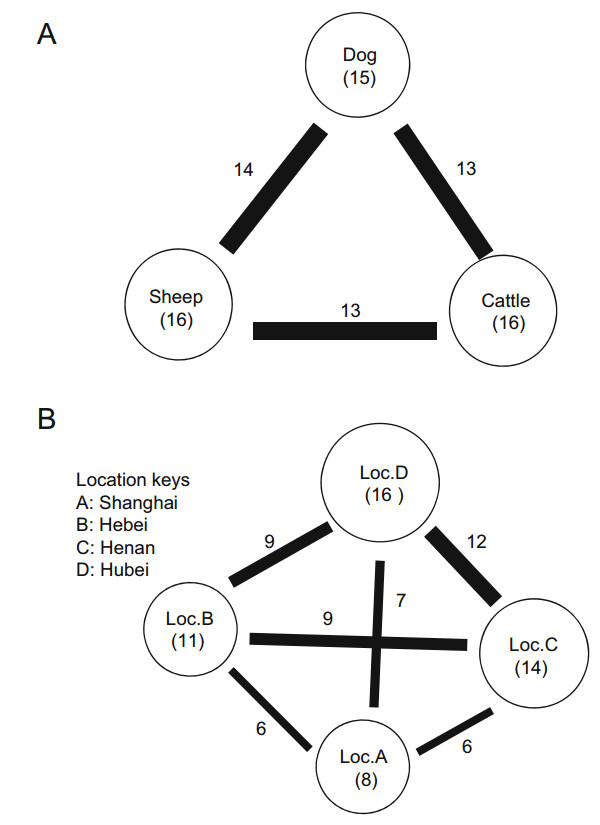
Figure 3. Virome similarities observed with different host species (A) and geographic locations (B). The size of the circle is proportional to the total number of viruses discovered in ticks sampled from each host species (A) or geographic location (B). Within each circle, information pertaining to the host species or geographic location and the number of viral families is provided in parentheses. The thickness of the line connecting the circles reflects the number of viral families shared between the host species or geographic locations. The number of shared viral families is shown next to the line.
Differences between the four locations were also reflected in the types of viral families that the ticks harbored (Fig. 3B). Although the tick pools from location C (Henan) and location D (Hubei) both contained more than 19 viral families, all were of low abundance. Conversely, location A (Shanghai) shared nearly all of the viruses identified in the other three locations. In particular, several novel viruses shared genetic identity with those found in a given species to a certain degree, including bole tick virus 3 (50%) and bole tick virus 1 (67%), and were mostly negative-sense RNA viruses.
-
To explore the diversity and putative evolutionary origin of the novel negative-sense RNA viruses identified in this study, a phylogenetic tree was established based on the sequence of the major capsid protein, VP1 (Fig. 4A). Clustering of tick-associated viruses from different countries or ticks species was apparent at many instances within the phylogenies, and in some cases, the monophyletic groups contained substantial genetic diversity suggestive of a long-term association between the viruses and their tick hosts. Notably, the tick-associated clusters often contained multiple viral lineages associated with single or multiple host species/genera, with no clear pattern of virus–host codivergence. The genome structures of the newly discovered viruses and their corresponding phylogenies are shown in Fig. 4B.

Figure 4. Evolutionary history and genomic features of NSRV1, which was discovered in this study. (A) The maximum-likelihood phylogenetic trees showed the position of NSRV1 (solid red circle) in the context of its closest relatives. The names of tick viruses identified in previous studies are marked in red, and the tick species from which they were identified are shown in square brackets. (B) The genome structures of the newly discovered viruses are shown for each corresponding genetic element. The predicted open reading frames in the genomes are labeled with information related to the potential protein or protein domain that they encoded.
We identified 6 putative RNA-virus families and some unclassified RNA viruses, representing all the major taxonomic categories (Table 3). Some of these RNA viruses were related to previously described tick viruses, whereas other viruses grouped with vertebrate viruses, plant viruses, fungal viruses, or other insect viruses, or had an uncertain host association. Studying the RNA-dependent RNA polymerase phylogeny showed that NSRV1 clustered within the most related viruses with a certain host association, whose host range was currently limited to arthropods. The closest relative of NSRV1 was bole tick virus 3. NSRV1 showed an L gene structure typical of bole tick virus 3, which has three substantially conserved sequences in the Mononegavirales RNA-dependent RNA polymerase, Mononegavirales mRNA-capping region V, and mRNA capping enzyme (Paramyxovirus family).
Negative-sense RNA virus 2 (NSRV2) mapped to a separate tick-associated cluster among different viruses with a certain tick species association. The closest relative of NRSV2 was found to be bole tick virus 1, which was initially identified in Hyalomma asiaticum in China. Although NSRV2 was not discovered in all six pools, it was of moderately high abundance (1344 hits) and clustered with viruses identified from the other tick hosts, suggesting that it potentially represents a novel tick-association virus from different tick species.
Finally, to summarize the potential host associations of all the viruses identified here, we considered several key attributes that were relevant to host association, including the abundance level, prevalence, and host association of close relatives. Among the 19 viral families identified here (Fig. 5), 6 were identified as RNA viruses and accounted for a large proportion in the virus hits.
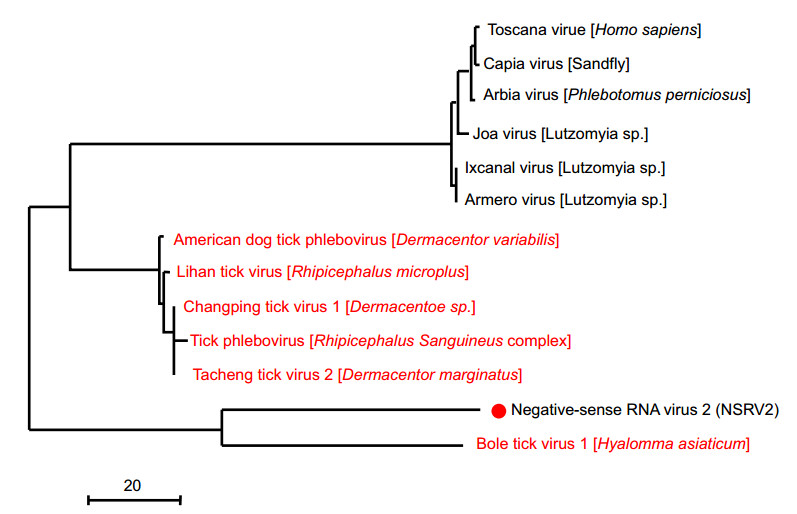
Figure 5. Evolutionary history and genomic features of negativesense RNA virus 2 (red dot), which was discovered in this study.
Tick Virome
Virome Ecology
Phylogenetic Analysis of the Newly Identified Negative-Sense RNA Viruses
-
In this study, we performed metagenomics analysis with ticks collected from eastern coastal areas and central areas of China because abundant species, large populations, and deaths related to tick-borne diseases have been reported for these regions (Tokarz et al. 2014). Our metagenomics approach generated 13, 721 high-quality reads that revealed key aspects of the ecology of RNA viruses in ticks from four sampling points. Based on updates made to the virus database, we included reference sequences to compare our results with those from previous studies (Manukian et al. 2006; Bolling et al. 2015; Li et al. 2015). Through a complex comparison process, we annotated 19 viral families, including 2319 sequencing reads annotated as vertebrate viruses, 11, 176 sequencing reads annotated as insect viruses, 72 sequencing reads annotated as plant viruses, and 62 sequencing reads annotated as phages.
The detection of different viromes in ticks from all four surveyed areas suggested that ticks from the eastern coast tolerated a higher diversity of viruses than did ticks from central areas. Similarly, among the previously described vector-borne viruses, little overlap was observed between the viruses carried by different vectors, such that ticks from the four locations harbored 8–19 of the same viral families in this study. Hence, the similarity in viromes among the five tick species may in part reflect their close evolutionary relationship, which may in turn dictate similarities in the cellular environment, immunological response, and perhaps ecological niche. The two tick genera also exhibit a large discrepancy in virus numbers and abundances, which is robust across all comparisons despite the relatively small sample size.
Ticks are known to harbor various pathogens, although research on the diversity of tick-associated viruses is limited (Rizzoli et al. 2014; Diuk-Wasser et al. 2016). In this study, Phenuiviridae/Phlebovirus, Retroviridae, and Caulimoviridae were found in all six tick pools. The Phlebovirus genus (Phenuiviridae family) is classified into the tick-borne viral groups and includes many viruses that threatens the health of humans and animals (Elliott and Brennan 2014; Alkan et al. 2016). Phlebovirus was found as a novel bunyavirus in China in 2011 and could be transmitted by Haemaphysalis and Rhipicephalus ticks (Yu et al. 2011).
In contrast to differences in viromes between genera, the Rhipicephalus virome was relatively homogenous among the species and across the regions sampled. Furthermore, some of the viruses identified here were found not only in the eastern coast and central areas of China, but also in regional countries like Turkey and America (Szabó et al. 2005; Aktas et al. 2006), indicating that they infect hosts over a wide geographical area. The viruses present in these different countries are relatively dissimilar (50%–64% nucleotide identities), and the observation of such genetic distances tentatively suggests that these viruses were introduced by windblown ticks or cyclones from neighboring regions, or that they were inadvertently spread by humans, rather than the result of ancient tick dispersal. In addition, we found that these viruses had shared similarity with known viruses, suggesting they could be classified as new viruses.
Finally, it is important to note that among the various virus species identified here, a small part of them fell into the category of "vector-borne" viruses that are known to infect humans or other mammalian hosts. Indeed, in a previous report, most of the tick viruses discovered either clustered with "arthropod-specific" viruses or were uncharacterized (Shi et al. 2015). These findings suggest that human and vertebrate pathogens represent only a tiny fraction of the tick virome, although it is possible that they exist at very low copy numbers if they exhibit low levels of replication. Whatever the cause, the observation that vector-borne viruses are rare further indicates that characterizing tick viromes provides important insight into the ecology and evolution of insect viruses.
This study has important limitations and implications. Due to the study design and collective evaluation of multiple tick specimens, the actual source of the identified sequences could not be determined precisely and might have been of environmental origin. Moreover, the potential health impacts of most of the identified viruses on human and animal are not currently known and must be elucidated. The MiSeq data were confirmed by performing polymerase chain reaction experiments, and more studies are needed to screen for these newly identified viruses.
This viral survey provides a baseline for comparing viruses detected in ticks, according to species, natural hosts, and geographic locations. The similar viral families detected in same tick genus and even ticks from different natural hosts indicate that commensal infections occur in the same tick genus and that, to some extent, the natural hosts were not the major deciding factor of the virus diversity. Furthermore, the geographic location was highly related to the viral abundance and diversity.
-
This work was supported by the National Key Research and Development Program of China (2017YFC1200202) and the Shanghai Pudong New Area Science and Technology Development Fund (Grant No. PKJ2018-N02).
-
HG and LC designed the experiments. TZ, XS, TS, XY, and SW carried out the experiments. TZ and WZ analyzed the data. XS and TZ wrote the paper. TZ and LC checked and finalized the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: