














HTML
-
ALT Alanine transaminase anti-HAV Antibody to hepatitis A virus anti-HBc Antibody to hepatitis B core antigen anti-HBe Antibody to hepatitis B e antigen anti-HBs Antibody to hepatitis B surface antigen anti-HCV Antibody to hepatitis C virus anti-HDV Antibody to hepatitis D virus anti-HEV Antibody to hepatitis E virus CA9 Carbonic anhydrase 9 CHB Chronic hepatitis B DNA Deoxyribonucleic acid DOCK8 Dedicator of cytokinesis 8 DP Double positive ELISA Enzyme-linked immunosorbent assay GWAS Genome wide association study HBeAg Hepatitis B e antigen HBsAg Hepatitis B surface antigen HBV Hepatitis B virus HBx Hepatitis B x protein HCC Hepatocellular carcinoma HDV Hepatitis D virus HEV Hepatitis E virus IgM Immunoglobulin M LLOD Lower limit of detection PCR Polymerase chain reaction SD Standard deviation SP Single positive TOF Time of flight mass spectrometry WES Whole exome sequencing
-
HBV infection is a serious worldwide public health issue. Despite the application of antiviral drugs such as nucleos(t)ide analogues, interferons (IFNs) and hepatitis B vaccine, there are still about ~257 million people worldwide who are chronic carriers of HBV (WHO 2017) and have a mortality risk from HBV-related liver diseases, such as liver cirrhosis, hepatic decompensation and hepatocellular carcinoma (Ding et al. 2016). In the natural course of HBV infection, less than 5% of cases in adulthood (WHO 2015) and 90% of neonates and 20%–60% of children under the age of 5 years fail to clear HBV and eventually develop into chronic HBV infection (Hoofnagle et al. 2007). The clinical manifestations of HBV infection vary among different infected subjects, ranging from acute self-limiting infection, inactive carrier state, fulminant hepatic failure to chronic hepatitis with potential diverse severe progression and complications (Wang et al. 2010). However, the factors that contribute to the persistence are unknown.
It was commonly believed that HBsAg and anti-HBs (protective antibody) seldom appear simultaneously in serum of the same patient. However, chronic HBV replication is able to occur despite the presence of anti-HBs in the serum of certain infected individuals. Coexistence of HBsAg and anti-HBs was first reported by Arnold in 1976 (Arnold et al. 1976), and later many other studies reported this phenomenon as well (Colson et al. 2007; Fu et al. 2017; Xiang et al. 2017). Chen et al. (Chen et al. 2011) reported significant higher aa substitution diversity within HBV viral S gene, especially the MHR and the "a" determinant in patients who display coexistence of HBsAg and anti-HBs, most frequently in the s126, s129 and s130 sites. Chen et al. (2011) also found that the incidence of basal core promoter double mutations (A1762T/G1764A) in the coexistence group was higher than in controls. Hadiji-Abbes et al. (Hadiji-Abbes et al. 2015) reported that the C69R variant contributes to a conformational change that occur not only at mutation site but also in the immunodominant "a" region, causing a decrease on the binding affinity to anti-HBs. In addition to the viral factors, host factors may also influence the presence of this concomitant situation. We previous reported that variants within the OAS3 gene was associated the presence of HBsAg and anti-HBs in patients with chronic HBV infection (Wang et al. 2018).
The pathogenesis and clinical manifestations of HBV infection are due to the interaction between virus replication and host immune responses to HBV-encoded antigens (Chisari et al. 2010). For viral factors, many studies had investigated virus genetic factors that may have influence on the progression of HBV infection. Researchers sequenced basal core promotor of HBV genome and found that patients with T1762/A1764 mutation were associated with progression of liver diseases, pathogenesis of HBV infection, and were more likely to develop HCC than controls (Kao et al. 2003). Several studies further confirmed that patients with T1762/A1764 mutations were associated with an increased risk of severe liver diseases including HCC (Kuang et al. 2004; Liu et al. 2006; Zheng et al. 2011). For host factors, Saxena et al. (2014) found that several genotypes in interleukin 6 (IL-6) (-572/-597) gene influence HBV disease progression. For example, the GA haplotype in -597G > A of IL-6 decreased the risk of hepatitis, cirrhosis, and subsequent HCC development among HBV carriers. Other cytokine genes such as interleukin-10 (IL-10), interleukin-28 (IL-28), transforming growth factor-β1 (TGF-β1), tumor necrosis factor-α (TNF-α) and migration-inhibitory factor (MIF) may also affect the susceptibility to HBV infection (Qi et al. 2009; Zhang et al. 2011; Zheng et al. 2012; Korachi et al. 2013; Zhang et al. 2013; Zeng 2014). Several studies revealed that HLA-DPA1-A and HLA-DPB1 alleles were associated with the viral clearance of HBV and HCC development (An et al. 2011; Hu et al. 2012). Earlier, Diepolder et al. (1998) found that patients with chronic hepatitis B (CHB) had lower HLA-DR13 allele than in healthy controls, suggesting that the HLA class Ⅱ allele DR13 was associated with self-limiting of HBV infection.
HBV infection development is a multifactorial process with implicated influences of both host and viral genetic determinants. Any factors that may influence the host's immunity towards hepatitis B virus are likely to affect the outcomes of HBV infection. However, previous studies mainly focused on the common host potential genetic factors, which may miss rare functional genetic variants. Our previous study (Wang et al. 2018) was focus on genetic factors that influence anti-HBs expression. To reveal additional associated variants, we conducted a two-stage GWAS-bioinformatic analyses aiming to find other potentially functional variants that influence HBV persistent infection.
-
We carried out a retrospective survey of HBV related diseases in Chinese Han population from May, 2002 to June, 2017 and conducted a study with two stages of bioinformatic analyses aiming to find the potential genes that may play a role in different status of HBV related diseases. In the first stage (Wang et al. 2018), 101 cases positive for both HBsAg and anti-HBs [double positive (DP)] and 102 control subjects who show positive for anti-HBs but negative for HBsAg [single positive (SP)] were included. All these subjects are age and gender matched and genotyped by whole exome sequencing (WES). For the second stage, we expanded our samples [579 cases who are chronic HBV infection and 439 controls who are HBV clearance (HBsAg negative but anti-HBs and anti-HBc positive)] and conducted a phenotypic analysis using time of flight mass spectrometry (TOF) to further validate the result of the first stage. All the samples included in this study were mainly obtained from Peking University First Hospital and the Fifth Hospital of Shijiazhuang. The inclusion and exclusion criteria applicable to all samples are listed in Table 1.
Inclusion criteria Cases (DP, chronic HBV infection)
1. HBsAg, anti-HBc positive for at least 6 months and no history of hepatitis B vaccination
2. anti-HAV, anti-HEV, HDAg negative and /or anti-HDV negative
3. Anti-HCV negative, HCV RNA negative
4. For DP cases in the first stage, anti-HBs positive for at least 6 months; for chronic HBV infection cases in the second stage, anti-HBs can be positive or negativeControls (SP, HBV clearance)
1. Anti-HBs and anti-HBc positive or anti-HBs positive and no history of hepatitis B vaccination; HBsAg negative
2. HBV-DNA negative, anti-HAV, anti-HEV, HDAg negative and /or anti-HDV negative
3. Anti-HCV negative, HCV RNA negativeExclusion criteria* 1. Evidence of past or current infection by HCV or HDV
2. With other hepatitis virus infection
3. Other systemic disease not related to HBV infection
4. Age less than 18 for all cases and controls
5. Not of Han ethnicityDP: double positive; SP: single positive.
*Excluded from enrollment if one or more of the exclusion criteria were met, applicable for all the two stage samples.Table 1. Inclusion and exclusion criteria.
Case definitions of different status of HBV infection are in consistent with the criteria issued by the Association of Infectious Diseases of China in 2015 (Hou and Lai 2015). In briefly, the cases of these two stages are chronic HBV infection, the controls are HBV clearance. The study was approved by the Ethics Committee of Peking University First Hospital and the Fifth Hospital of Shijiazhuang. Before entering this research group, all the subjects had signed an informed consent.
-
Virological and serological tests were processed at local sites. Serum HBsAg, anti-HBs, HBeAg, and anti-HCV were detected using the ARCHITECT I2000 test (Abbot, USA). HBsAg higher than 0.05 IU/mL and anti-HBs higher than 10 mIU/mL were defined as positive, respectively. HBV DNA was quantified using Roche Cobas Ampliprep/Cobas Taqman PCR with lower limit of detection (LLOD) of 20 IU/mL (Roche, USA) or commercial real-time polymerase chain reaction kit with LLOD of 100IU/mL (Daan Company, China). Anti-HAV IgM, HDV antigen, anti-HDV and anti-HEV were determined by commercially ELISA kits in China.
-
Genomic DNA of host was extracted from peripheral blood clot using protocols of QIAamp DNA Blood Mini Kit (QIAGEN) or salting-out method conducted by Tianyi Huiyuan Company (http://www.dna1953.com.cn/index.html). After that, 2-3 micrograms of DNA per individual from the first stage of 101 cases and 102 controls were delivered to BGI (http://www.genomics.cn/index) and were then sequenced and analyzed using WES on Illumina Hiseq X-Ten. Detailed procedure can be seen in our published article (Wang et al. 2018). The quality of exon sequencing was strictly controlled to guarantee that > 80% of the targets were covered by at least 20×. Reads containing adapters, uncertain bases (N) > 10% or low-quality bases (Phred < 5) > 50% were filtered out and the remaining clean sequence reads were then compared to the human reference genome hg19 (http://genome.ucsc.edu/, build 37.1) using BWA (http://bio-bwa.sourceforge.net/index.shtml, 0.7.15). In addition, the duplicate fragments were labeled using Picard (http://picard.sourceforge.net/).
-
After base quality recalibration and local realignment around the potential Indel sites, the Genome Analysis Toolkit (GATK, http://www.broadinstitute.org/gatk/index.php, v3.6) was applied to call SNPs and Indels. Then VEP (http://grch37.ensembl.org/info/docs/tools/vep/index.html, release-77) and ExAC databases were used to conduct annotation, which provide information such as mutation allele frequency, gene variant consequences and altered function of protein.
-
After alignment, mutation detection and annotation of the clean data, we conducted further filtering to identify high-confidence variants in targeted sequence. Variants meeting all the following requirements were considered to be high-confident: (1) quality (QUAL) ≥ 100; (2) depth of coverage ≥ 6 and support variant reads ≥ 3; (3) pass the allele balance test (prop test P > 0.0005); (4) the interval of two variants > 5bp. High-confidence variants of all samples were merged to one Variant Call Format (VCF) file using bcftools. The call rate threshold of final VCF file was set to 80% and variants that failed to achieve the threshold were excluded from this GWAS dataset. After that, a genetic association analysis was carried out with fisher's exact test between cases and controls in the first stage.
-
TOF (Griffin et al. 1999) is based on single base extension molecular reaction, different allele has different molecular weight and different flying time in the electric field, which makes it possible to classify. TOF is the most important technical platform for stage Ⅱ study by GWAS, it can effectively avoid the false-positivity caused by traditional fluorescence signal technique, which can ensure more accurate and correct results. To further determine the potential gene variants found in the first stage of WES, we performed TOF among larger samples which composed of 579 cases with chronic HBV infection and 439 controls who are HBV clearance with anti-HBs and anti-HBc positive but HBsAg negative. Genomic DNA of host was extracted as mentioned above. We applied iPLEX GOLD (Sequenom MassARRAY) to design primers for PCR and the PCR products were then digested by SAP enzyme to remove free dNTPs from the system. After that, we had a single base extension reaction. Then we transferred the purified products to the 384-well Spectro-CHIP bio-array to conduct analysis using MALDI-TOF mass spectrometer. The raw data and genotype map were obtained by using TYPER4.0 software. After checking the completeness and correctness, the results were stored in appropriate storage media and submitted to the biological information room for analysis.
-
In the first stage of WES analysis, we chose the first 150 potentially functional variations (missense mutation, frameshift mutation, stop-gained mutation and stop-lost mutation) ranked by P value. To further confirm the discovery-phase genetic factors, we searched the related genes of these 150 variations in databases (such as PubMed, EMbase, Cochrane) and chose those that may have a relationship with HBV related liver diseases. Combining consideration of primer design in the process of TOF, we selected 30 variants from the above 150 loci as candidate genes and analyzed whether there was genetic discrepancy between cases and controls.
-
Regarding to the demographic and clinical characteristics of the subjects included in this study, Chi-square test and t-test was applied using SPSS17.0 statistical software. Single variant association analysis was conducted by PLINK 1.07 software using Fisher's exact test.
Study Design and Population
Laboratory Examination
Library Preparation and Whole Exome Sequencing
SNP and Small Indel Detection
GWAS Dataset Construction
Genetic Factors Confirmation by Time of Flight Mass Spectrometry (TOF)
SNP Selection in Replication Study
Statistical analysis
-
Characteristics of participants in the first stage were delineated in our previously published article (Wang et al. 2018).
Characteristics of the 1018 subjects in the second stage who received a TOF analysis were described in Table 2. Individuals in the control group were significantly older than those in the case group (P < 0.05), whereas gender was well balanced between the two groups (P > 0.05). Compared to the control group, ALT, HBV DNA levels and HBeAg positive rate in the case group were significant higher (P < 0.05).
Group Cases (n=579) Controls (n=439) P value Age, y Mean (x ± SD) 49.57±14.74 (579) 61.93±13.40 (439) 0.000 Range 18-84 18-96 Male, %(n) 55% (317/579) 59% (261/439) 0.142 HBsAg positive, %(n) 100% (579) 0 (439) 0.000 anti-HBs positive, %(n) 6.8% (39/571) 100% (439) 0.000 HBeAg positive, %(n) 18% (103/565) 0 (0/439) 0.000 ALT, IU/mL, mean±SD (n) 32.74±60.06 (456) 21.88±23.86 (333) 0.000 HBV-DNA, log IU/mL, mean±SD (n) 1.63±2.51 (277) 0.00±0.00 (14) 0.000 P values less than 0.05 are indicated in bold
ALT Alanine transaminase; anti-HBs antibody to the hepatitis B surface antigen; DP double positive; HBeAg hepatitis B e antigen; HBsAg hepatitis B surface antigen; SD standard deviationTable 2. Characteristics of subjects in the second stage of time of flight mass spectrometry.
-
We performed a genome-wide association study of 58, 336 polymorphism variants that have available authoritative transcript using Fisher's exact test in all samples of 101 cases and 102 controls and conducted a Bonferroni correction (significance level was set as P < 0.05/58336) because of the existing multiple tests. However, no loci achieved this significance threshold. Fig. 1 delineated the P value of all these single variants. The first 150 potentially functional variations ranked by P value are displayed in Supplementary Table S1. We also showed the characteristics of published CHB risk-associated SNPs (Chang et al. 2014) in Supplementary Table S2. However, those loci are not in the exome capture region, so the performance are unavailable in our first stage of WES.
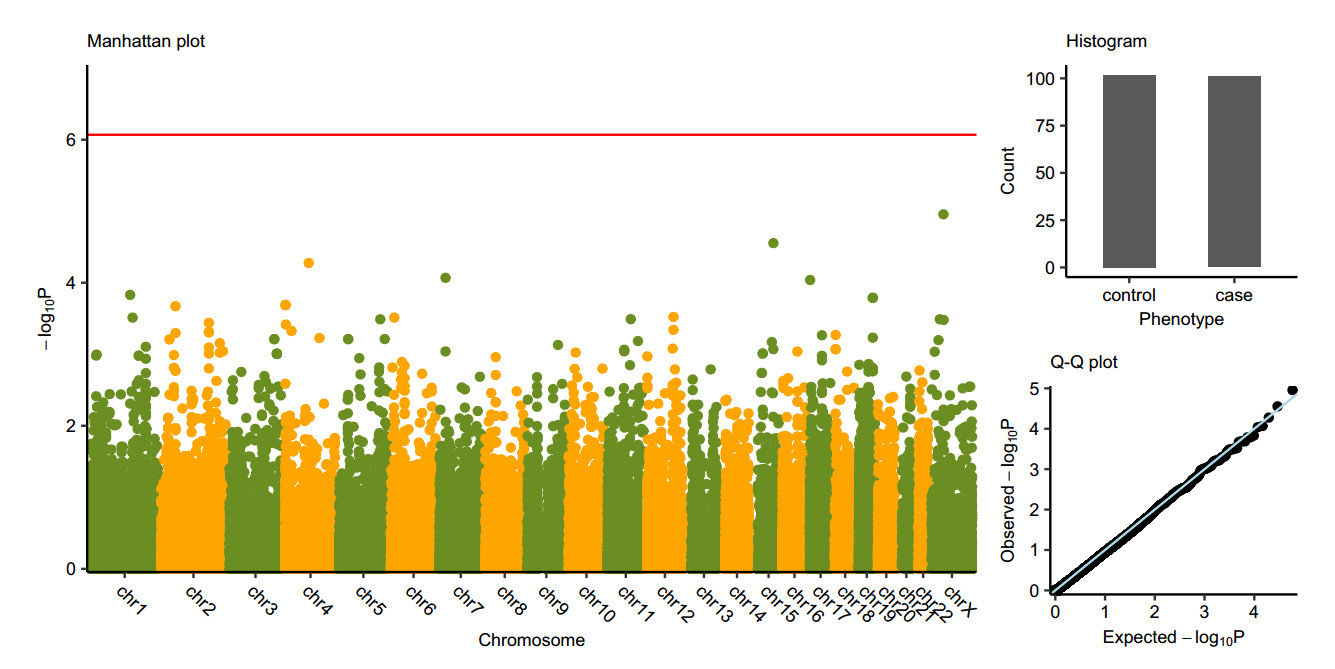
Figure 1. Manhattan plot
-
In the first stage GWAS, we failed to detect any potentially functional association. In view of this, we expanded our sample sizes and performed this second stage (candidate gene analysis) using TOF in 30 leading associated variants to reveal an association with chronic HBV infection (Table 3). There were several sites that achieved significant difference, including rs11040923, rs2071676, rs2288868, rs4774113 and rs506121. SNPs rs2288868 and rs4774113 were excluded due to Hardy-Weinberg Equilibrium departure (P < 0.05) and < 95% call rate in the cases group respectively. SNP rs11040923 was also excluded due to discordant allele frequencies in stage 1 vs stage 2 populations.
Position Case (n%) Control (n%) OR (95%CI) P value rs1048906 62.37 64.45 0.91 (0.76–1.10) (C/T)* 0.35 rs10821128 46.37 49.20 0.89 (0.75–1.06) (C/T) 0.21 rs11040923 67.00 62.4 1.22 (1.02–1.47) (A/G) 0.035 rs16932912 34.30 35.10 0.97 (0.80–1.16) (A/G) 0.74 rs17206365 70.80 71.60 0.96 (0.79–1.18) (A/T) 0.72 rs1870134 28.60 26.10 1.14 (0.93–1.39) (C/G) 0.21 rs2071676 52.60 46.70 1.27 (1.06–1.51) (A/G) 0.009 rs2073674 55.10 55.70 0.98 (0.82–1.16) (A/C) 0.79 rs2075688 100 100 - (C) - rs2272662 52.80 55.90 0.88 (0.74–1.05) (C/T) 0.19 rs2277603 77.90 81.30 0.81 (0.65–1.01) (A/G) 0.06 rs2288868 80.60 72.50 1.58 (1.28–1.94) (C/T) 2.28E-5 rs2297879 45.20 49.20 0.85 (0.71–1.01) (C/T) 0.07 rs2302061 29.70 29.00 1.03 (0.85–1.25) (C/T) 0.77 rs3732487 46.00 49.00 0.89 (0.75–1.06) (G/T) 0.19 rs3733662 28.70 31.40 0.88 (0.73–1.06) (A/C) 0.19 rs3745535 34.60 36.10 0.94 (0.78–1.13) (A/C) 0.51 rs3779234 76.70 76.90 0.99 (0.80–1.22) (C/T) 0.92 rs3804769 16.30 17.10 0.94 (0.75–1.19) (C/T) 0.63 rs3815045 23.90 22.50 1.08 (0.88–1.33) (A/G) 0.49 rs3818123 46.30 48.20 0.93 (0.78–1.11) (C/T) 0.42 rs4629585 46.40 47.70 0.95 (0.79–1.13) (A/C) 0.56 rs4774113 17.60 21.40 0.79 (0.63–0.99) (G/T) 0.04 rs4938941 27.90 25.70 1.11 (0.91–1.36) (A/G) 0.29 rs506121 60.30 65.10 0.81 (0.68–0.97) (C/T) 0.027 rs553717 69.40 68.20 1.06 (0.88–1.28) (C/T) 0.56 rs723077 54.20 53.50 1.03 (0.86–1.23) (A/C) 0.75 rs760749 78.20 76.10 1.13 (0.92–1.39) (A/C) 0.26 rs8100856 44.90 45.10 0.99 (0.83–1.18) (C/T) 0.96 rs934945 71.80 75.10 0.85 (0.70–1.04) (C/T) 0.106 P values less than 0.05 are indicated in bold
OR Odds ratio; CI confidence interval.
*If the OR was calculated as C/T, then the frequency listed in the table is the frequency of C among all the subjectsTable 3. Alleles discrepancy of the selected 30 variants between cases and controls in the second stage of time of flight mass spectrometry.
Polymorphisms Stage Case (n%) Control (n%) OR (95%CI) P value rs506121 (DOCK8) 1 55.90 68.10 0.59 (0.40–0.89) (C/T)* 0.014 2 60.30 65.10 0.81 (0.68–0.97) (C/T) 0.027 1+2 (meta) 0.77 (0.65–0.91) (C/T) 0.002 rs2071676 (CA9) 1 58.90 43.60 1.85 (1.25–2.75) (A/G) 0.002 2 52.60 46.70 1.27 (1.06–1.51) (A/G) 0.009 1+2 (meta) 1.35 (1.15–1.58) (A/G) 0.0003 rs11040923 (DNHD1) 1 63.40 77.00 0.52 (0.34–0.80) (A/G) 0.003 2 67.00 62.40 1.22 (1.02–1.47) (A/G) 0.035 1+2 (meta) 1.07 (0.91–1.27) (A/G) 0.40 P values less than 0.05 in meta-analysis are indicated in bold
DOCK8 Dedicator of cytokinesis 8; CA9 carbonic anhydrase 9; DNHD1 dynein heavy chain domain 1; OR odds ratio; CI confidence interval.
*If the OR was calculated as C/T, then the frequency listed in the table is the frequency of C among all the subjectsTable 4. Allele discrepancy of rs506121, rs2071676, rs11040923 between cases and controls.
Table 4 delineates allele discrepancies of rs506121, rs2071676, rs11040923 between cases and controls in the two stages. Patients who were HBsAg positive in the case group had elevated DOCK8 –T allele (rs506121; P < 0.05) and CA9 -A allele (rs2071676; P < 0.05) than that in clearance group in both stages. A meta-analysis to evaluate the performance of these three SNPs (Table 4), which suggested a similar result for SNPs rs506121 and rs2071676 (P = 0.002, OR=0.77, 95%CI [0.65, 0.91]; P = 0.0003, OR=1.35, 95%CI [1.15, 1.58], respectively).
Characteristics of Participants in This Study
Genetic Analysis between Groups in the First Stage of WES
Genetic Analysis between Groups in the Second Stage of TOF
-
The outcomes of HBV infection can vary depending on differing viral and host genetic determinants factors. Previous studies of host genes mainly focused on common genes such as cytokines and HLA genes, however, rare gene may have an influence as well. Based on our previous study that mutation of OAS3 gene may affect the presence of anti-HBs in patients with chronic HBV infection (Wang et al. 2018), we enlarged sample size and focus on persistent HBV infection compared to HBV clearance to reveal new potential variants that may have an effect on the clearance or persistence of HBV infection.
In the first stage, we did not find any potentially functional gene variants that may have a relationship with HBV infection at the Bonferroni genome-wide significance level (Fig. 1). Reasons accounting for this may be as follows: first, exome sequencing only focuses on genetic variation in the exome region, and is thus very likely to neglect those lying in the noncoding region (Wang et al. 2018); second, statistical power of classical single-variant based association tests for low-frequency and rare variants is low when the sample sizes are small (Lee et al. 2014; Wang et al. 2018). In the following stage, we expanded our sample sizes and further conducted a TOF analysis to confirm the potential gene variants found in the discovery-phase. Two-stage design is the standard strategy of genome-wide association study for discovering and validating the candidate genetic makers. Stage 2 is a replication study which focuses on the statistical significance discovered in stage 1. Previous study demonstrated that joint analysis of replication-based study result in increased power to detect genetic association (Skol et al. 2006). And for evaluating the significance of association between genetic factors and affected-unaffected phenotypes, Chi-square test, Fisher's exact test (Purcell et al. 2007) and mixed regression model (Zhou et al. 2012) are the most popular methods designed for population matched and un-matched study respectively. In our study, we used Chi-square test to perform the joint analysis on 30 variants in two populations. Combining the results of these two stages, we observed that cases in CHB group have significantly higher frequency of DOCK8 -T allele and higher frequency of CA9 -A allele than controls in clearance group (Table 4).
Autosomal recessive mutations in the DOCK8 gene, located on chromosome 9p24.3, can result in DOCK8 immunodeficiency (Betts et al. 2015), a syndrome predisposing carriers, to various viral or fungal infections (Zhang et al. 2009). DOCK8 is a member of the DOCK180 superfamily of atypical guanine-nucleotide exchange factors (GEF) (Cote et al. 2002) and can activate cell division cycle 42 (Cdc42) (Mizesko et al. 2013). Previously, Nishikimi et al. concluded that Cdc42 is a member of Rho GTPase, which functions as molecular switches by cycling between an inactive GDP-bound state and an active GTP-bound state (Nishikimi et al. 2013) and is important for reorganization of filamentous actin (F-actin) cytoskeleton in natural killer (NK) cells and dendritic cells (DCs) (Sinai et al. 2010; Harada et al. 2012). The loss of DOCK8 in mice may lead to DCs' less competitive to migrate from peripheral tissues to lymph nodes and act its role as an antigen-presenting cell, which being able to initiate an adaptive immune response (Krishnaswamy et al. 2015). Some studies demonstrated that there exhibits a similar phenomenon of a migration defect of macrophages and CD8+ T cells in the absence of DOCK8 (Kearney et al. 2017; Shiraishi et al. 2017), being unable to perform their function as clearing pathogen. It was previously described by some researchers that patients with DOCK8 deficiency exhibit an impaired T-cell, B-cell, NKT cell and RORγt+ innate lymphoid cells (ILCs) function, which may affect the persistence of these cells (Boztug et al. 2012; Kearney et al. 2017). Jabara et al. concluded in their study that DOCK8 acts as an adaptor, which can associate toll like receptor 9 (TLR9) with Pyk2-Src-Syk-Stat3 signaling cascade via Myeloid differentiation primary-response protein 88 (MyD88) and that the lack of DOCK8 may result in TLR9 mis-localization, crippling the ability of TLR9 to activate Stat3 and disabling subsequent TLR9-driven B cell activation, proliferation and immunoglobulin production, which will impair the humoral response mediated by B cells (Jabara et al. 2012). Pei et al. (2014) reported that MyD88 plays a part in the signaling cascade of the innate immune response mediated by TLRs. Further, overexpression of MyD88 in liver cells or mouse model can inhibit HBV replication via downregulation of viral transcription or acceleration of the decay of viral pre-genomic RNA (Xiong et al. 2004; Li et al. 2010). Many studies had previously described that TLRs signaling pathways play an essential role in the control of HBV replication (Isogawa et al. 2005; Pei et al. 2014) by modulating the expression of microRNAs (Zhang et al. 2011; Li et al. 2013) or inducing antiviral cytokines (Isogawa et al. 2005). In a case-control study, researchers found TLR mutations in rs3804099 and rs4696480 were significantly associated with milder hepatitis activity among patients with chronic HBV infection, concluding that the activation of TLR pathways may further intensify the inflammation of hepatocytes (Lin et al. 2018). Ligands of TLR2, TLR3, TLR4, TLR7, and TLR9 in hepatoma cells have been reported to inhibit HBV replication (Xia et al. 2008). Taking the discoveries of previous studies and the results of our current study, we speculate that DOCK8 -T allele may increase persistence of HBV infection thereby impairing the immune system, TLRs function and the correlated signaling pathways.
Carbonic anhydrase (CA) is a family of zinc metalloenzymes and can efficiently catalyze the reversible processes of hydration-dehydration of CO2 and HCO3-, which can help maintain the neutral pH in hypoxic condition. Hypoxia is commonly observed in tumor microenvironment and can lead to various genetic and adoptive responses that accommodates tumor growth (Yoo et al. 2010). Hua et al. (2017) suggested that CA9 variants pose a prognostic marker of hepatocellular carcinoma (HCC), and that CA9 allele regulate tumor growth and metastasis of HCC cells through controlling cell proliferation and mobility and the epithelial-mesenchymal transition (EMT) process in HCC cells. Hyuga et al. (2017) suggested that CA9 variants are associated with poor prognosis through regulation of the EMT process in HCC. Kang et al. (2015) reported in two cohorts that those with high CA9-positive had poorer prognosis after surgery for HCC than those with low CA9-positive, which suggested that CA9 expression may be a prognostic factor for survival. Holotnakova et al. (2010) demonstrated that HBx increased the CA9 promoter activity and that HBx involves a portion of HBV DNA integrated into hepatocyte chromosomes, which explains the role of HBx in HBV infection with HCC. Holotnakova et al. (2010) also hypothesized that CA9 could enhance the hepatocyte tolerance to ischemia, which can increase the viability of HBV infected hepatocytes and assist further development such as oncogenesis. Lastly, Wang et al. (2013) found variants of CA9 gene may change the host susceptibility to certain tumors leading to different outcomes.
In this study, we found the CA9 -A allele to be elevated in chronic HBV infection (Table 4). CA9 is located on chromosome 9p12-13 and contains 11 exons, which can encode for the 459-amino-acid protein (Wang et al. 2013). Mutation at locus rs2071676 of CA9 gene results in the alteration of amino acid (NM_001216:c.G96A; p.Val33Met), which may affect the function of this gene-encoded protein and mRNA conformation and thereafter have an impact on disease (Shen et al. 2018). We proposed that rs2071676 variants of CA9 gene might also be a risk factor for the chronicity of HBV infection, in a way mediated by possible alteration of protein function.
We conducted a two-stage analysis and demonstrated that SNP alleles of DOCK8 and CA9 gene may increase the risk of chronicity of HBV infection, which may serve as a marker for the management of HBV infection. Further functional studies are needed to measure the expression of these two gene-coded proteins in HBV-related diseases to help management of HBV infection.
-
This study was supported by grant from the International Science & Technology Cooperation Program of China (No.2014DFR31200), the National Infrastructure of Chinese Genetic Resources (YCZYPT [2017]01-6), federal funds from the National Cancer Institute, National Institutes of Health, USA (No. N01-CO-12400), and the National Natural Science Foundation of China (No. 30671855).
-
ZZ, BBW and JGZ designed the study. MJF, SW, JW, TYL, HP, ZRF, LC, EHD, JHL, HLX and YYY collected samples and analyzed data. MJF, SW, JW, JGZ, BBW and ZZ analyzed and interpreted the data with the assistance of HKL, HFX, DVZ, and SJO. MJF and JW wrote the manuscript. ZZ revised and approved the final manuscript. All authors had full access to the final version of the report and agreed to the submission.
-
The authors declare that they have no conflict of interest.
-
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and with 1964 Helsinki declaration and later amendments or comparable ethical standards. The study was approved by the Ethics Committee of Peking University First Hospital and the Fifth Hospital of Shijiazhuang. Before entering this research group, all the subjects had signed an informed consent. This study does not contain any studies with animals performed by any of the authors.

 DownLoad:
DownLoad: