














-
Dear Editor,
African swine fever (ASF) is one of the most important porcine infectious diseases and has caused huge economic losses worldwide (Teklue et al. 2020). African swine fever virus (ASFV), the causative agent of ASF, is a double stranded DNA virus with a genome about 170 to 194 kb in length, and encodes 150 to 167 open reading frames (ORFs). There are more than 50 proteins involved in the mature virions. Among these proteins, p72 encoded by B646L gene is the essential component in the formation of ASFV particles. The sequence of B646L gene is also usually used for the genotyping of ASFV, and there are 24 B646L genotypes (Quembo et al. 2018).
Since the first report in Kenya (Montgomery 1921), ASF has been introduced into dozens of countries across three continents (Africa, Europe and Asia) in the nearly 100 years (Teklue et al. 2020). In August 2018, the first outbreak of ASF in China was confirmed in Liaoning Province (Zhou et al. 2018), and more than 180 cases has been reported in China by July 2020 (https://www.oie.int/wahis_2/public/wahid.php/Countryinformation/Countryreports).
ASFV causes a mortality rate up to 100%, and could interfere with various cellular signaling pathways and further affect the immunomodulation (Revilla et al. 2018). So far, no effective vaccines against ASFV are available. Thus, timely and accurate diagnostic tools, which could be used on site, are urgently needed to control the spread of ASFV. The gold standard of ASFV diagnosis recom-mended by World Organization for Animal Health (OIE) is virus isolation (OIE 2019). However, virus isolation is time-consuming and is not suitable for on-site diagnosis.
Several polymerase chain reaction (PCR) assays, including conventional PCR and quantitative real-time PCR (qPCR) assays, have been developed for ASFV detection, and these PCR assays were the most frequently used routine diagnostic methods in laboratories (Oura et al. 2013; Gallardo et al. 2019). However, the conduct of PCR assays requires sophisticated laboratory equipment, and the observation of conventional PCR products requires electrophoresis, making PCR assays unsuitable for on-site diagnosis. Isothermal amplification techniques amplifies DNA under isothermal conditions, that is, only simple devices are needed, such as a water bath or a heat block (Notomi et al. 2000). Loop-mediated isothermal amplification (LAMP) is a well-known isothermal amplification technique, and its four to six primers ensure it could amplify target DNA with high specificity and sensitivity (Becherer et al. 2020). In previous studies, two LAMP assays, targeting topoisomerase II gene and p10 gene, have been developed for ASFV detection with the detection limits of 330 and 30 copies per reaction, respectively (James et al. 2010; Wang et al. 2020). Mehran Khan and colleagues have showed that the sensitivity of LAMP is higher than that of qPCR (Khan et al. 2017). But a qPCR assay targeting the ASFV B646L gene possessed a detection limit of 18 DNA copies (Fernandez-Pinero et al. 2013). These means the LAMP method for ASFV diagnosis could be improved. Moreover, LAMP combining with lateral flow dipstick (LFD) could visualize the prod-ucts by the naked eye without affecting its sensitivity (Zhang et al. 2014). In this study, LAMP combined with LFD (LAMP-LFD), targeting the ASFV B646L gene, was developed for on-site diagnosis of ASFV. PCR assays, including conventional PCR, qPCR and nested PCR (nPCR), and LAMP monitored by electrophoresis targeting the same conservative region of B646L gene were devel-oped for comparison.
Primers for PCR and LAMP assays (Supplementary Table S1) were designed based on the conservative region of B646L gene of 42 ASFV strains (Supplementary Figure S1), and operation details of these assays were presented in Supplementary Materials. Eight ASFV strains distributed in 4 clades (Supplementary Figure S1) were picked up for specificity study, and the sequences of B646L gene of them were artificially synthesized by Sangon Biotech (Shanghai, China). In addition, the genome sequence of one porcine circovirus (PCV) strain was also artificially synthesized for the specificity study. BamHI and SalI (Takara, Beijing, China) cutting sites were added separately on the 5' - and 3' -end of the artificially synthe-sized sequences. Subsequently, the sequences were individually cloned into the pEASY® -T1 vectors (Transgen, Beijing, China). Other swine viruses (Supplementary Table S2), including 6 RNA viruses (classical swine fever virus (CSFV), group A rotaviruses (RVA), porcine epidemic diarrhea virus (PEDV), transmissible gastroenteritis virus (TGEV), porcine reproductive and respiratory syndrome virus (PRRSV) and deltacoronavirus (delta-CoV)) and 2 DNA viruses (pseudorabies virus (PRV) and porcine parvovirus (PPV)), were stored at -80 ℃ LC and used in the specificity study. All the ASFV clinical samples were firstly inactivated in a BSL-3 laboratory, and then viral DNA was prepared in a BLS-2 laboratory, and other viruses used in this study were also inactivated in a BLS-2 laboratory.
Under optimized conditions, all the assays produced corresponding positive products or signal. Conventional PCR and nPCR produced specific bands with expected lengths in agarose gels (Fig. 1A). qPCR assay generated amplification curve in ASFV positive reaction (Fig. 1B), and according to the standard curve, Ct values (y) and log of copy numbers (x) were linearly correlated (y = - 3.504x + 39.978, E = 92.9%, R2 = 0.998), the melting curve showed a single peak indicating that no primer dimers were formed (Fig. 1C). The positive LAMP reaction produced symbolic ladder-like bands in 2% agarose gel (Fig. 1D). In LAMP-LFD assay, positive reaction generated clearly visible red line on the positions of test line and control line (Fig. 1D). No false-positive results were observed in the negatives control reactions of all the assays. The specificity study showed that all the assays yielded positive results or signals when ASFV templates existed in the reaction mixtures (Fig. 1E). While, no false-positive results were observed when testing 9 other swine virus templates. These results indicated that the primers used in this study were highly specific to ASFV templates.
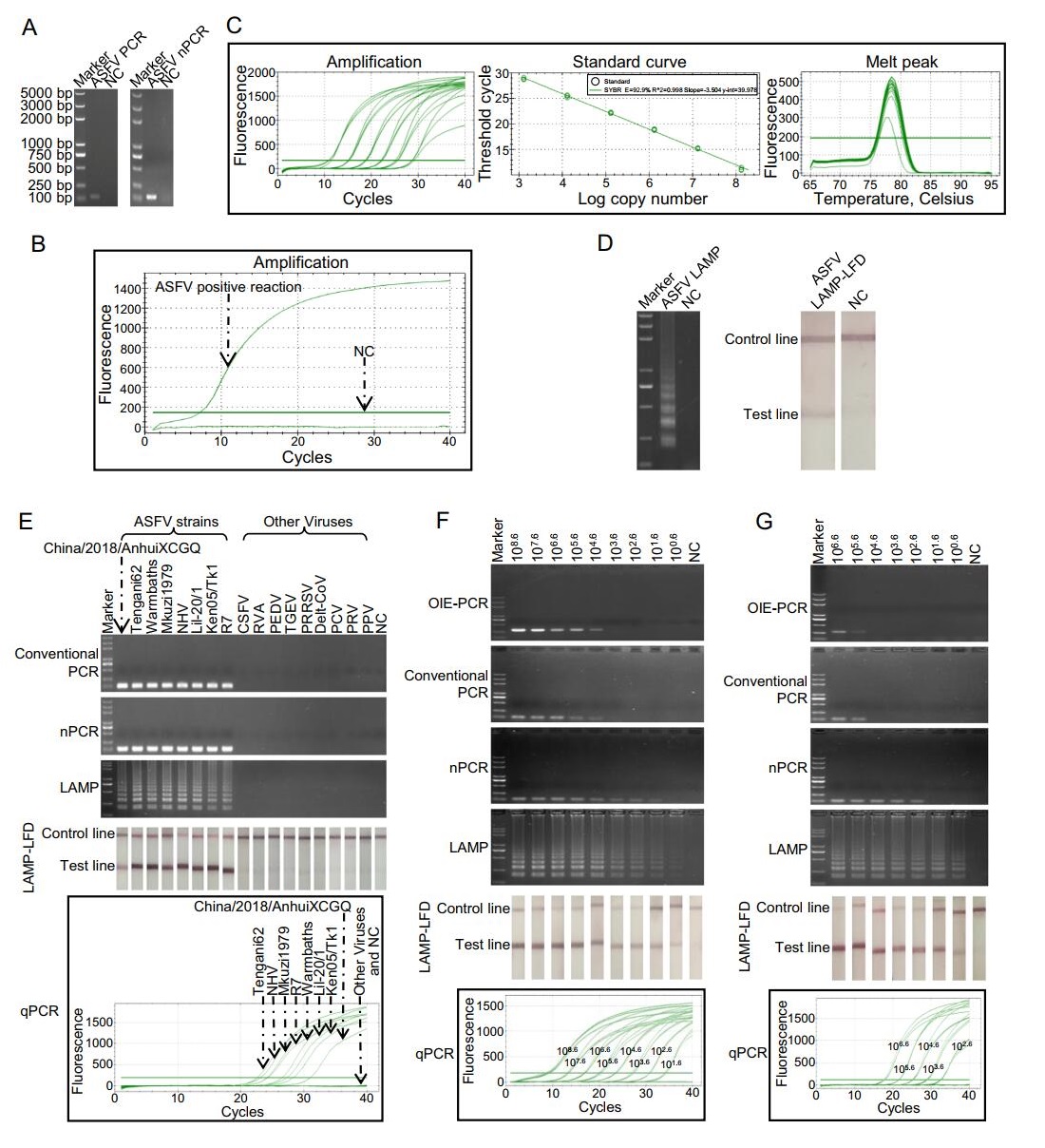
Figure 1. Comparison of specificity and sensitivity of PCR and LAMP assays for ASFV detection. A Products of conventional PCR and nPCR in agarose gel. B Amplification signal of qPCR reaction. C The standard curve of ASFV qPCR. The standard curve was generated by plotting the CT (y) values vs. log copy numbers (x). The reaction efficiency (E) of qPCR was 92.9%. y = -3.504x + 39.978. D The products of LAMP monitored by electrophoresis and LFD. E Specificity study of PCR and LAMP assays. F Sensitivity study of PCR and LAMP assays detecting purified B646L fragments. G Sensitivity study of PCR and LAMP assays detecting mimics of ASFV-infected tissue. Markers in the electrophoresis were all the same and its molecular weights were labeled on the left of (A). In (A-B and D-F), ddH2O was used as template in negative control reaction (NC). In (G), DNA extracted from healthy spleen was used as template in NC. In qPCR, copy numbers of templates used in each reaction were labeled on the amplification curves.
For sensitivity study, 10-fold serially diluted purified B646L fragments prepared by digested pEASY® -T1 vectors carrying B646L fragments with BamHI and SalI were used, and an OIE-recommended conventional PCR (OIE-PCR) was set as a reference method (Aguero et al. 2003). The detection limit of OIE-PCR was 104.6 copies/reaction. The minimum detection abilities of conventional PCR, nPCR, qPCR, LAMP and LAMP-LFD were individually 104.6, 101.6, 101.6, 100.6 and 100.6 copies/reaction (Fig. 1F). Our conventional PCR had the same detection limit as OIE-PCR, showing that the quality of new designed pri-mers was good. Consistent with previous study (Zhang et al. 2014), LAMP monitored by electrophoresis and LAMP-LFD showed identical sensitivity. Moreover, in line with the result of Khan and colleagues who showed LAMP assay was more sensitive than qPCR in Phytophthora infestans diagnostic (Khan et al. 2017), our result revealed that LAMP method was 10 times more sensitive than qPCR in B646L fragments detection.
Additionally, to evaluate the ability and the detection limits of the assays in clinical samples, mimics of ASFV-infected tissue were formed by mixing B646L fragments with spleen taken from healthy pig. When detecting these mimics, the lowest detection limits of the OIE-PCR, conventional PCR, nPCR, qPCR, LAMP and LAMP-LFD were 105.6, 105.6, 102.6, 102.6, 100.6 and 100.6 copies/reaction, respectively (Fig. 1G). Additionally, DNAs from 52 clinical samples (collected and treated by Sichuan Provincial Center for Animal Disease Control and Prevention accordance with the standard operation for ASFV) were used for testing the utility of the assays. LAMP and LAMP-LFD showed the highest positive rate (16/52), and the positive ratios in OIE-PCR, PCR, nPCR and qPCR were 13/52, 13/52, 14/52 and 15/52, respectively. LAMP and LAMP-LFD showed higher sensitivity in ASFV-infected mimics testing and one more positive sample in clinical samples, this may be because LAMP assays were more tolerant to the presence of non-target tissue DNA that could disturb the amplification of the PCR assays (Notomi et al. 2000; Inacio et al. 2008). The one more positive sample in clinical samples examination was from a positive farm which has been judged according other samples through qPCR. The controversial sample could not be further verified because the samples were inactivated. More detections of clinical samples are needed to examine that LAMP could do better that qPCR but not false positive.
Altogether, we have developed LAMP-LFD as a timely and accurate detection tool for on-site ASFV detection. By comparison with PCR assays, including conventional PCR, qPCR and nPCR, the LAMP-LFD assay showed high specificity and sensitivity. Our data indicate that LAMP-LFD is a promising method for ASFV diagnosis in the field, and it's even applicable in some low-resource locations.
HTML
-
This research was funded by Key R & D Program of Sichuan Province (2018NZ0151) and Major Science and Tech-nology Special Program of Sichuan Province (2018NZDZX0006).
-
We declare that we have no conflict of interest.
-
The animal experiment in this study was approved by the Animal Ethics Committee (ACE) of Sichuan University (license: SYXK-Chuan-2018-185). All experiment proce-dures and animal welfare standards strictly followed the guidelines of Animal Management at Sichuan University.

 DownLoad:
DownLoad: