












HTML
-
Coronaviruses (CoVs) are enveloped, positive-sense and single-stranded RNA viruses that infect humans and animals, which can cause enteric, hepatic and neurological diseases (Wang et al. 2015; Shi et al. 2016; Ge et al. 2017). Several of them are notorious etiological agents and have leaded to epidemic, such as SARS-CoV and MERS-CoV (Drosten et al. 2003; Zaki et al. 2012). In 2020, CoVs have aroused unprecedented public concern due to the ongoing COVID-19 pandemic caused by SARS-CoV-2 (Phan et al. 2020; Wu et al. 2020; Zhou et al. 2020). Due to the unique viral replication process, CoVs have a high mutation rate and recombination frequency (Lai and Cavanagh 1997). These features increase the possibility for CoVs to adapt to novel hosts and environments (Herrewegh et al. 1998; Woo et al. 2006; Lau et al. 2012, 2016; Moreno et al. 2017), having a potential of avian-to-mammalian and avian-to-avian transmission (Lau et al. 2018).
CoVs belonging to the family Coronaviridae were divided into four genera: Alpha-CoV, Beta-CoV, Gamma-CoV and Delta-CoV (https://talk.ictvonline.org/taxonomy/). Generally, bats are the reservoirs of Alpha-CoV and Beta-CoV, and birds are the reservoirs of Gamma-CoV and Delta-CoV (Woo et al. 2012; Lau et al. 2015; Xu et al. 2016). The genus Delta-CoV contains seven species (https://talk.ictvonline.org/taxonomy/) from both mammals and birds (Woo et al. 2012; Lau et al. 2018). Porcine CoV HKU15, now commonly referred to as porcine deltacoronavirus (PDCoV), has been identified as one of the major enteric pathogen to pigs causing diarrhea accompanied by vomiting, dehydration, loss of weight and death (Wang et al. 2014; Chen et al. 2015; Jung et al. 2015; Ma et al. 2015). Together with PDCoV, sparrow CoV (SpCoV) HKU17 and four SpCoVs (ISU690-4, ISU690-7 ISU42824 and ISU73347) belong to the species of Coronavirus HKU15 in genus Delta-CoV (Chen et al. 2018; Lau et al. 2018).
Marmot (Marmota himalayana) was identified as a major host of Yersinia pestis in Qinghai-Tibet plateau of China (Ge et al. 2015) and has been continuously monitored under China's Plague Surveillance Program. Several of our recent studies have found the possibility of marmots to be the host and reservoir for other pathogens including a new subtype of tick-borne encephalitis virus and novel bi-segmented and unsegmented picobirnaviruses (Dai et al. 2018; Luo et al. 2018).
Considering the highly-adaptive and potential pathogenic nature of coronaviruses, we collected samples from various wild animals from Yushu Tibetan Autonomous Prefecture, Qinghai Province, where has the greatest biodiversity among the world's high-altitude regions. According to the next-generation sequencing data and phylogenetic analysis, we identified a novel delta-CoV from wildlife samples and a novel host Montifringilla taczanowskii.
-
In July 2019, 29 marmots and 50 rats were obtained from Yushu Tibetan Autonomous Prefecture in Qinghai Province of China, and corresponding faecal samples were collected. Twenty-five birds near the plateau pika holes from different sites were also obtained while acquiring plateau pikas. Their faecal samples were collected. All the samples were preserved in viral transport medium (Lau et al. 2015) and transferred to our lab in Beijing by cold chain transportation and stored at −80 ℃. Marmots and rats were identified by professionals through morphological observation. Mitochondrial cytochrome b (Cyt b) gene was used to identify bird species (Sorenson et al. 1999; Saetre et al. 2001). All sampling work was conducted as part of plague surveillance in animals carried out by Yushu Prefecture Center for Disease Control and Prevention.
-
The RNA was extracted from each faecal sample using QIAamp viral RNA mini kit (Qiagen, Germany). Total RNA from rats, marmots and birds was grouped into three separate libraries for Illumina HiSeq2000 sequencing. After removing adapters, low-quality reads and host/rRNA sequences, high-quality (clean) data was applied to de novo assembly using Trinity version 2.4.0 (Grabherr et al. 2011) and annotated using BLASTx search in non-redundant protein database. To verify the assembled genomes, reads of clean data were mapped back to the obtained almost complete genomes of coronaviruses using Hisat2 version 2.1 (Kim et al. 2015), respectively.
-
To screen the prevalence of coronaviruses in corresponding samples, the 954 bp fragment of delta-CoV obtained in this study (delta-F1: GCTACGGAACGACCTGGGAT; delta-R1: ATTGGTTTGCGTCTGAGGTGA) and 440 bp fragment of beta-CoV (beta-F1: GGTTGGGATTATCCTAAGTGCGA; beta-R1: ACCATCATCACTCAAAATCATCA) based on RNA-dependent RNA polymerase gene (RdRp) were respectively amplified by PrimeScript™ One Step RT-PCR Kit Ver.2 (Takara, Japan). PCR products were examined using 1.5% agarose electrophoresis and Sanger sequenced.
-
Sequences were aligned using Mafft version 7 (Katoh and Standley 2013). Based on Akaike information criterion 1 (AIC1), best fit nucleotide substitution models of genome (GTR + I + G), OFR1ab (LG + G + F), S (WAG + G + F), M (LG + G) and N (LG + G + F) analyses were conducted using Model Generator version 0.57 (Keane et al. 2006). Phylogenetic trees were reconstructed using PhyML 3.0 (Guindon et al. 2010) and visualized in Tree of Life version 1.0 (Letunic and Bork 2016).
-
Genome sequences of thrush coronavirus (ThCoV) HKU12, munia coronavirus (MunCoV) HKU13, SpCoV ISU690-4 and Montifringilla taczanowskii CoV HM (obtained in this study, as query sequence) were aligned using Mafft version 7 (Katoh and Standley 2013). Bootscan analysis in SimPlot version 3.5.1 (Lole et al. 1999) was used to detected the possible recombination events. Parameters were set as step 200 bp, window size 1, 000 bp and model F84.
-
The RdRp nucleotide sequences of genus delta-CoV were aligned. Divergence dates were estimated using BEAST version 1.10.4 (Suchard et al. 2018). The most recent common ancestor (tMRCA) was calculated using GTR + G + I substitution model and uncorrelated relaxed clock type with log-normal relaxed distribution. The Bayesian Markov chain Monte Carlo was run for 2 × 108 generations with a sampling frequency of every 1, 000 steps. The ESS values of parameters should be greater than 200 and visualized using Tracer version 1.7.1. The tMRCA tree was annotated using TreeAnnotator version 1.10.4 in BEAST and visualized in FigTree version 1.4.4 (Suchard et al. 2018).
-
The genomes of CoVs obtained in this study were deposited in GenBank with accession numbers: MT215337, MT215336 and MT430884, respectively.
Sample Collection
RNA Extraction and Next-Generation Sequencing
Prevalence Screening of Identified Coronavirus in Animal Samples
Phylogenetic Analysis
Recombination Analysis
Divergence Dates Analysis
Nucleotide Sequence Accession Numbers
-
One full-length genome (31, 270 bp) related to beta-CoV was acquired from the pool of rats. To verify the assembled contig, a total of 64, 612 reads (0.62%) were mapped to the beta-CoV genome. The identified genome (named as Apodemus peninsulae CoV, accession numbers: MT430884) shared 94.5%–95.3% nucleotide (nt) identities with members of the species of Coronavirus HKU24. There was over 90% amino acid similarity regarding potential structural and non-structural proteins between Apodemus peninsulae CoV and closely related CoVs. To screen the prevalence of Apodemus peninsulae CoV in samples, we amplified the 440 bp fragment and the results showed that three out of fifty (6.0%) rat samples were positive (Table 1). For positive samples, two of them were Apodemus peninsulae and the other one was Microtus gregalis.
Name Accession number Sample types No. of positive samples/no. of test samples (% positive) Detected coronaviruses Apodemus peninsulae CoV MT430884 Rats 3/50 (6.0) Betacoronavirus MtCoV N MT215336 Birds 4/25 (16.0) Deltacoronavirus MtCoV HM MT215337 Marmots 1/29 (3.4) Deltacoronavirus Table 1. Prevalence of beta- and delta-CoV in animal samples collected in Qinghai-Tibetan Plateau in July 2019.
Two mostly identical genomes were identified to be related to delta-CoV. They were from the marmot pool and the bird pool, which were named as Montifringilla taczanowskii CoV (MtCoV) HM (MT215337) and MtCoV N (MT215336), respectively. The numbers of reads mapped to genomes of MtCoV HM and MtCoV N were 18, 676 (0.01%) and 10, 854 (0.01%), respectively. The prevalence of MtCoV in marmots and birds were 3.4% (1/29) and 16.0% (4/25), respectively (Table 1). Among positive samples, the four birds were Montifringilla taczanowskii, and the marmot was Marmota himalayana.
-
The two delta-CoV genomes were of the same length of 25, 896 nt with 41.3% G + C content and only had four nucleotide differences between each other at locations 2, 254, 5, 761, 10, 826 and 19, 787. The first two base changes (2, 254 and 5, 761) were synonymous changes, the third was in a noncoding region and the last (19, 787) was a nonsynonymous change in the protein spike (proline in MtCoV HM and serine in MtCoV N). The genome structure of MtCoV HM and MtCoV N (Fig. 1) shared high similarity with those of PDCoV and SpCoV HKU17 (Woo et al. 2012), including 5′ UTR (untranslated region), replicase ORF1ab, spike (S), envelope (E), membrane (M), nonstructural protein 6 (NS6), nucleocapsid (N), NS7a, NS7b, NS7c, and 3′ UTR (Fig. 1). The putative transcription regulatory sequence (TRS) was identified based on the motif 5′-ACACCA-3′ (Table 2). Interestingly, the distance between the TRS and the first base of the initiation codon of ORF NS7a is 101 bp, which is the longest compared with those of six members of the genus Delta-CoV that contained a NS7a gene (Woo et al. 2012), which ranged from 4 to 80 bp.
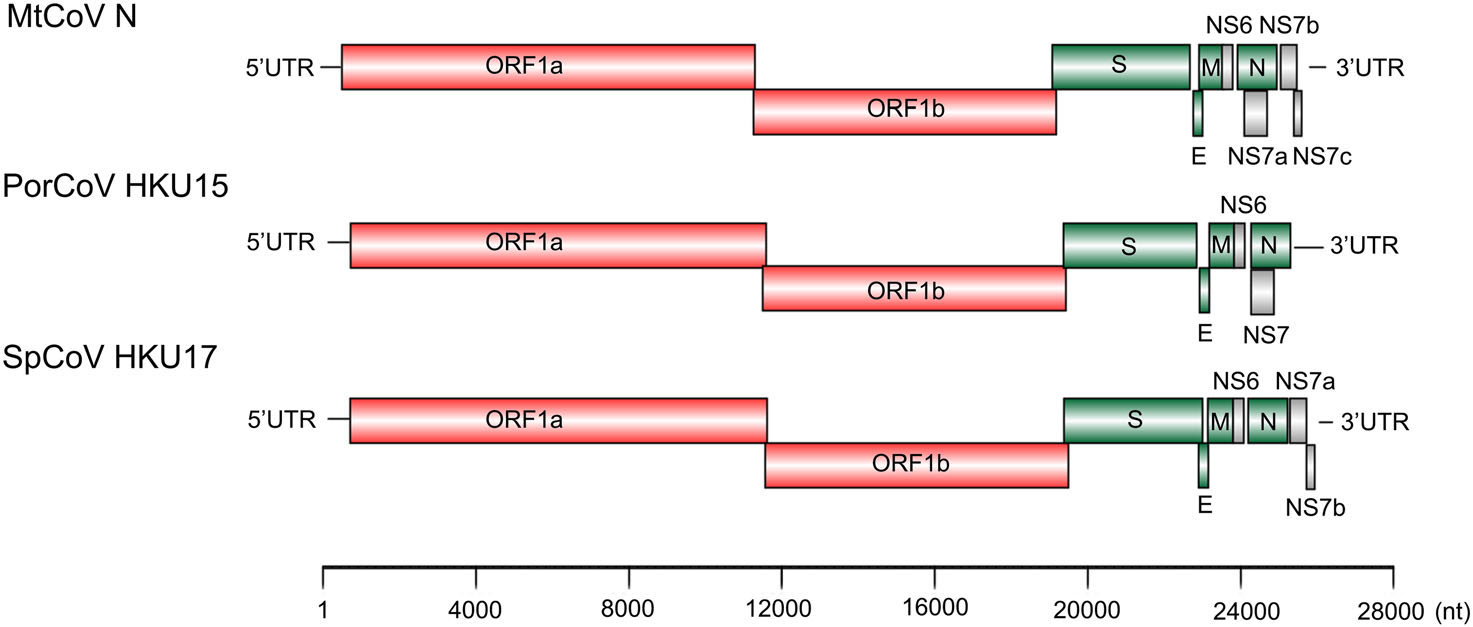
Figure 1. Comparision of genome organization of MtCoV N, PorCoV HKU15, and SpCoV HKU17.
MtCoV Location (nt) Length (aa) Frame(s) TRS location TRS sequence distance bases to AUG ORF1ab 497–19, 227 6, 242 +2, +1 37 ACACCA(453)AUG S 19, 209–22, 808 1, 199 +3 19, 062 ACACCA(140)AUG E 22, 802–23, 053 83 +2 22, 775 ACCCCA(20)AUG M 23, 046–23, 699 217 +3 23, 019 ACACCA(20)AUG NS6 23, 699–23, 980 93 +2 23, 646 ACACCA(46)AUG N 24, 005–25, 033 342 +2 23, 991 ACACCA(7)AUG NS7a 24, 099–24, 695 198 +3 23, 991 ACACCA(101)AUG NS7b 25, 044–25, 472 142 +3 25, 033 ACACCA(4)AUG NS7c 25, 394–25, 588 64 +2 25, 348 ACACGA(39)AUG Table 2. Potential codings and predicted transcription regulatory sequences of the genome of MtCoV.
MtCoV was identified as a novel member of the species. Pairwise nucleotide sequence alignment of the novel MtCoV genome showed the highest homologies to SpCoV ISU690-4 (83.3%), followed by SpCoV HKU17 (83.0%), QuaCoV UAE-HKU30 (78.5%) and PorCoV HKU15 (82.8%). The amino acid identities of ADRP, 3CLpro, RdRp, Hel, ExoN, NendoU and O-MT between MtCoV and their closely related strains were summarized in Table 3. Results showed that the concatenated seven replicase domains revealed more than 90% amino acid identity to the members of species of Coronavirus HKU15 (Table 3), which suggested that MtCoV belongs to this species (Lau et al. 2018). However, the structural proteins E, M and N in MtCoV showed lower identities (83.1%–84.3%, 86.6%–87.1% and 88.6%–90.4%, respectively) to SpCoV HKU17, SpCoV ISU690-4, QuaCoV UAE-HKU30 and PorCoV HKU15. In particular, protein S in MtCoV shared the highest amino acid identities to SpCoV HKU17 (73.1%), following with Houbara coronavirus (HouCoV) UAE-HKU28 (72.2%), Pigeon coronavirus (PiCoV) UAE-HKU29 (72.1%) and Falcon coronavirus (FalCoV) UAE-HKU27 (72.1%), but very low identities to other members of the same species (44.8%–45.4%). Overall, these lower identities of structural proteins between MtCoV and other members of the species of Coronavirus HKU15 indicated that MtCoV represents a novel member in that species.
Domain SpCoV HKU17 PorCoV HKU15 SpCoV ISU690-4 QuaCoV UAE-HKU30 Amino acid identity (%) MtCoV ADRP 94.5 95.3 93.8 89.8 3CLpro 90.8 88.6 91.5 89.9 RdRp 95.0 94.9 94.7 93.2 Hel 97.8 97.3 97.7 97.5 ExoN 95.9 94.8 95.4 94.6 NendoU 90.5 88.4 89.6 89.9 O-MT 91.4 90.3 92.5 92.8 Concatenated 94.5 93.7 94.3 93.4 S 73.1 44.8 45.3 45.4 E 84.3 83.1 83.1 83.1 M 87.1 87.1 86.6 87.1 N 90.4 88.9 89.8 88.6 Table 3. Comparison of amino acid identities between MtCoV and closely related CoVs.
-
Phylogenetic analysis of whole-genome nucleotide sequences showed Apodemus peninsulae CoV was closely related to the cluster including members of the species of Coronavirus HKU24 (Fig. 2). Also, the result further confirmed that MtCoV belongs to the genus Delta-CoV and forms an independent lineage. It was closely related to PorCoV HKU15 and SpCoV HKU17 (Fig. 2). Further, phylogenetic analyses based on amino acid sequences of proteins ORF1ab, M and N were identical to trees based on nucleotide sequences, and both revealed that MtCoV HM and MtCoV N were clustered with members of the species of Coronavirus HKU15 but in the meantime different from them (Fig. 3). The protein S based phylogenetic tree showed that MtCoVs were grouped with SpCoV HKU17, and were more closely related to FalCoV UAE-HKU27, PiCoV UAE-HKU29, HouCoV UAE-HKU28 and magpie robin coronavirus (MRCoV) HKU18 (Fig. 3). It was due to the high identities of protein S and was consistent with previous reports (Woo et al. 2012; Chen et al. 2018).
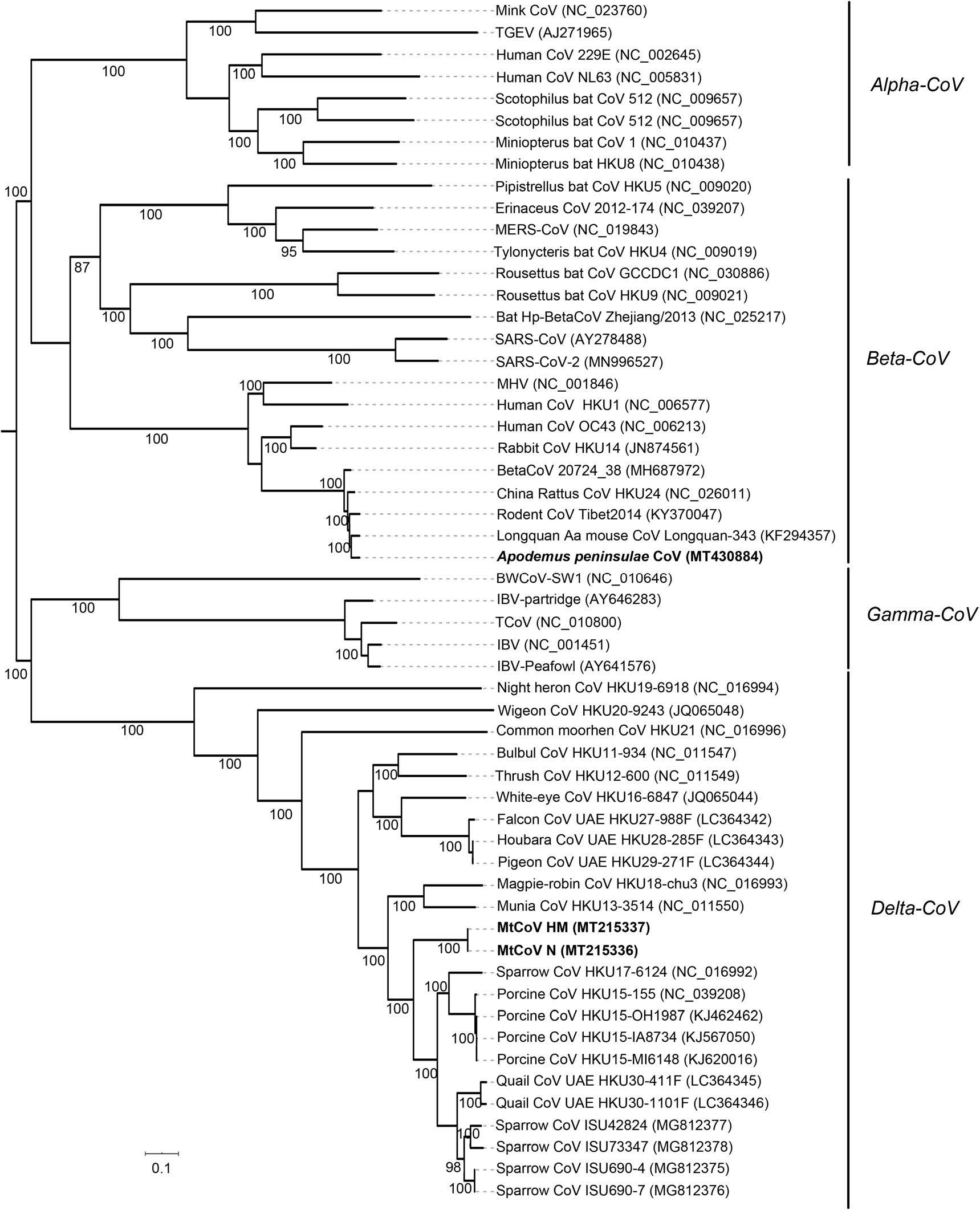
Figure 2. Phylogenetic analysis of genome sequences of coronaviruses. Bootstrap values (≥70%) are showed along branches. Scale bar suggests nucleotide substitutions per site.
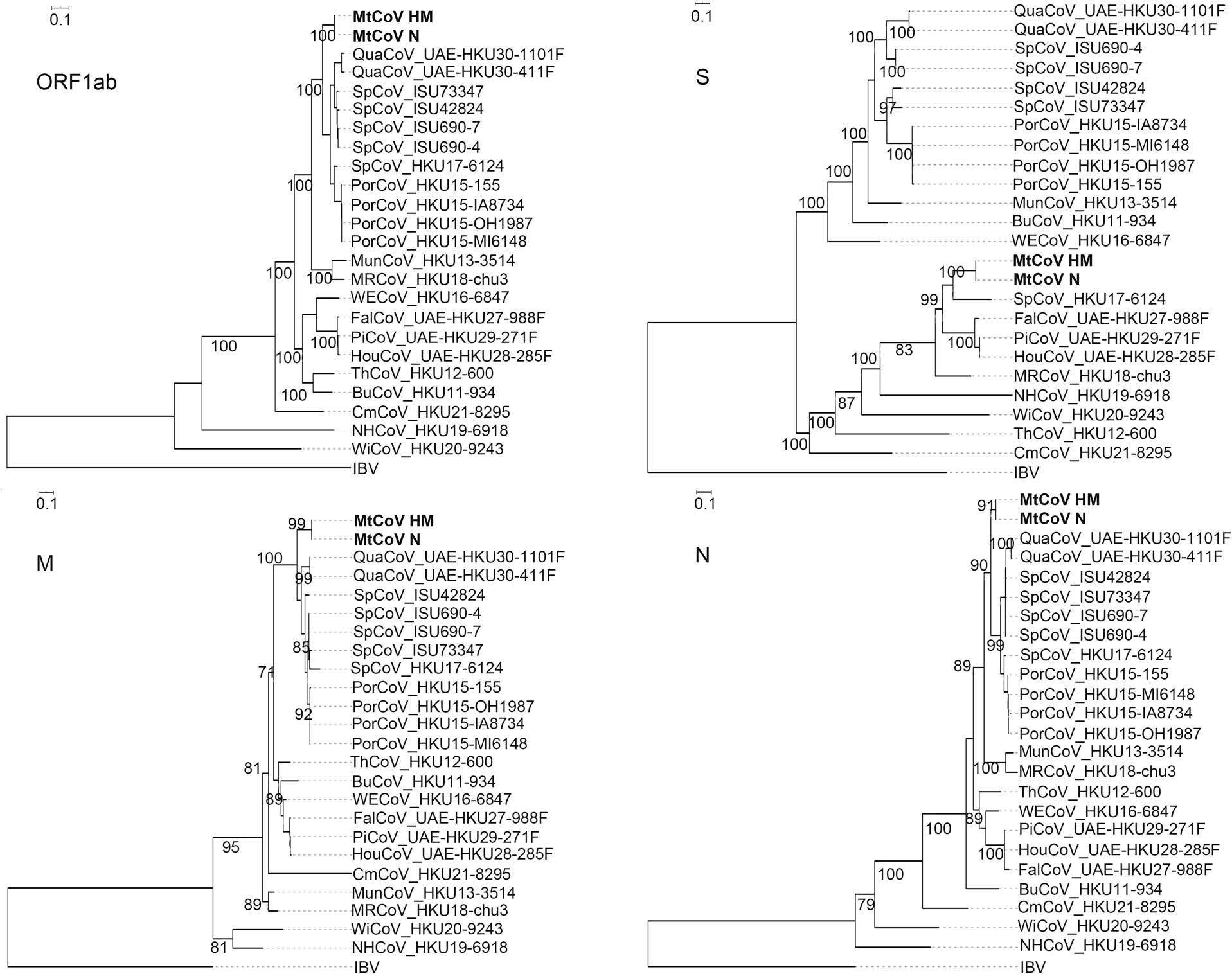
Figure 3. Phylogenetic tree analyses based on amino acid sequences of ORF1ab, S, M and N of coronaviruses. Bootstrap values (≥70%) are showed along branches. Scale bar suggests nucleotide substitutions per site. Bold strains are the novel ones isolated in this study.
-
The genomes of ThCoV HKU12, MunCoV HKU13, SpCoV ISU690-4 and MtCoV HM (as query sequence) were aligned for recombination analysis using Bootscan. The result indicated the potential long recombination segment from aligned positions 19, 500 to 23, 300, which were mainly located in the S gene of MtCoV (Fig. 4A). The recombinant segment was likely to be derived from ThCoV HKU12. Since the receptor-binding domain (RBD) locates in the S protein, the recombined sequence might lead to biological changes of receptor binding and thus initiate cross-species transmission. Two short potential recombination segments were also found in aligned positions 11, 300 to 11, 700 and 15, 500 to 16, 100 of MtCoV (Fig. 4A). Genome sequence of MtCoV HM (as the query sequence) was compared with that of ThCoV-HKU12 using Simplot analysis (Fig. 4B). Result from global align in BLAST indicated that nucleotide identity values of S gene between MtCoV HM (19, 209 to 22, 808 nt) and ThCoV-HKU12 (19, 433 to 23, 011 nt) was 58.5%, with part of S gene of MtCoVs showed above 70% nucleotide identity to ThCoV-HKU12 (Fig. 4B). And, S gene of MtCoVs shared higher nucleotide identity with PiCoV UAE-HKU29 (73.3%), HouCoV UAE-HKU28 (73.3%), FalCoV UAEHKU27 (73.2%), SpCoV HKU17 (71.0%), Magpie robin coronavirus HKU18 (67.4%), Night heron coronavirus HKU19 (58.7%) and Wigeon coronavirus HKU20 (58.6%).
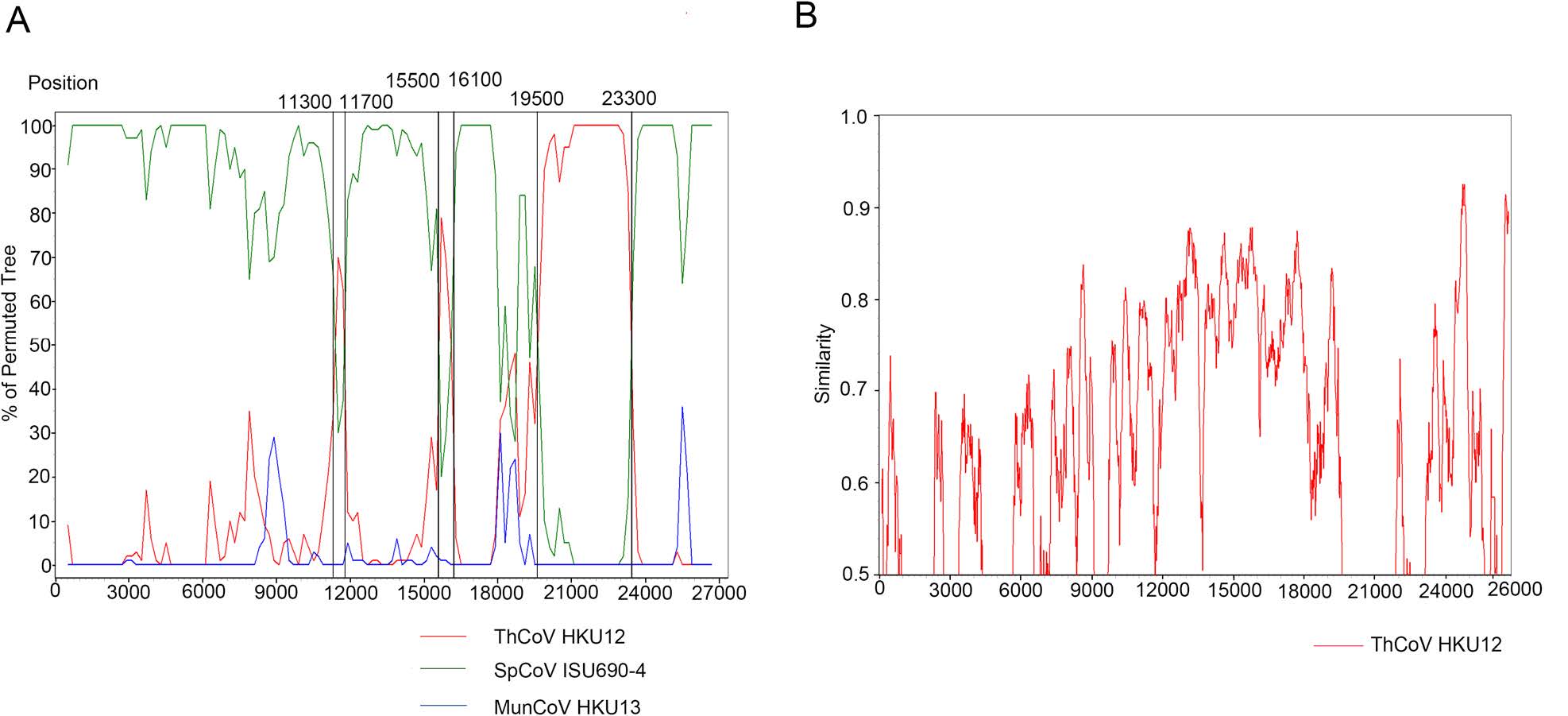
Figure 4. Potential recombination event detected using bootscan analysis. A Genome of MtCoV HM was used as the query sequence and compared with ThCoV HKU12, MunCoV HKU13 and SpCoV ISU690-4. Red lines indicated the recombination sites. B MtCoV HM was used as the query sequence and compared with the genome of ThCoV-HKU12.
-
To estimate the divergence time of MtCoV, the RdRp nucleotide sequences of genus delta-CoV were selected (Lau et al. 2018) and aligned to calculate tMRCA using GTR + G + I substitution model and uncorrelated relaxed clock type with the log-normal relaxed distribution. The result of molecular clock analysis indicated that the most recent common ancestor of MtCoVs along with other closest members of the species in Coronavirus HKU15 was estimated to be 289 (95% HPD = 60–1140) years ago (Fig. 5).
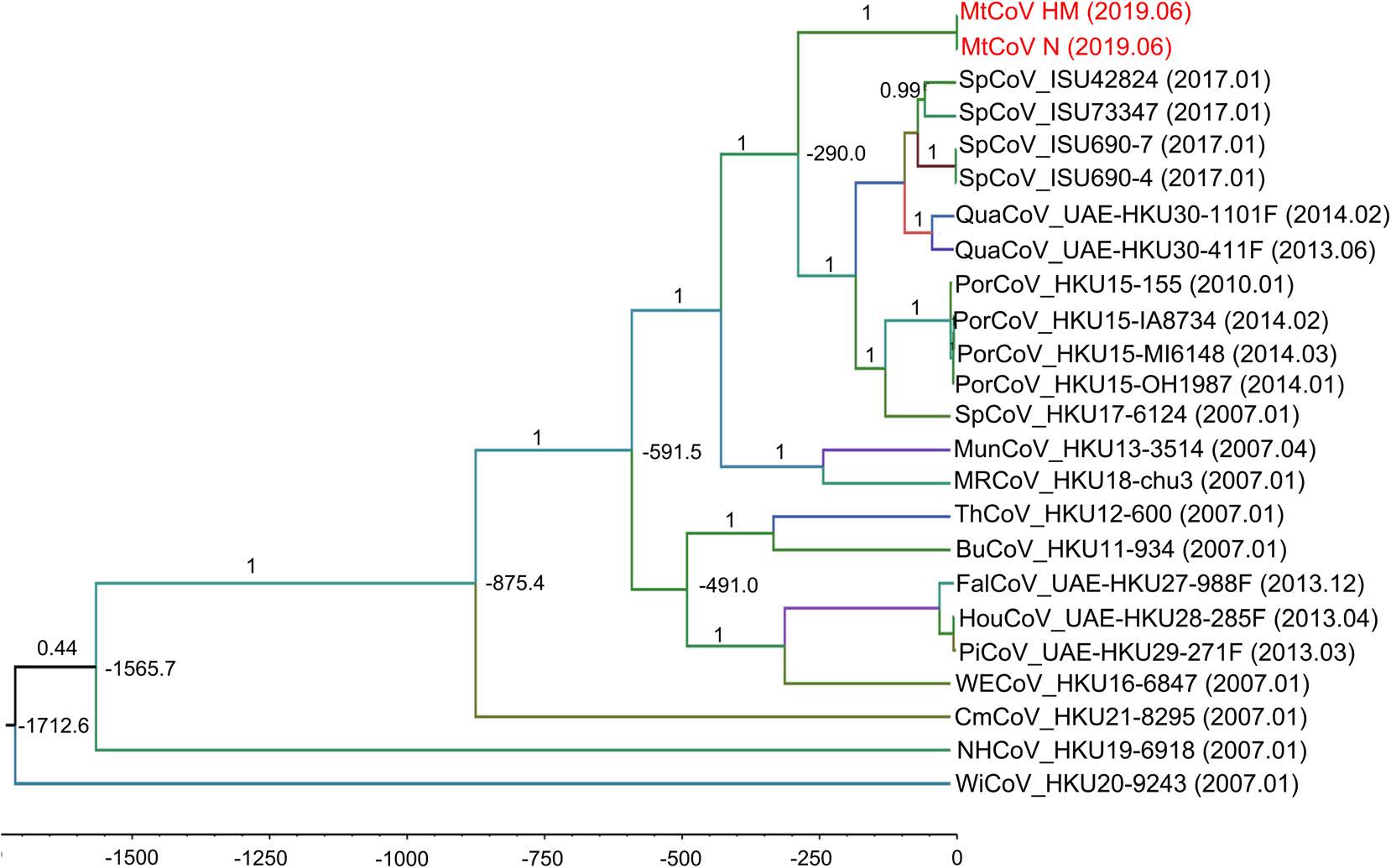
Figure 5. Bayesian Markov chain Monte Carlo (MCMC) tree analysis of the novel delta-CoV based on RdRp nucleotide sequences. The posterior probabilities and tMRCA are showed on branch labels and node labels. Sampling date is marked on the end of tip labels.
Identification of Beta-CoV and the Novel Delta-CoV
Genome Characterization of the Novel Delta-CoV
Phylogenetic Analyses of the Novel Delta-CoV
Recombination Analysis of Delta-CoV
Estimation of Divergence Dates of Delta-CoV
-
Based on the genome data, we report the characterization of two coronaviruses identified from the wild animal samples in Yushu Tibetan Autonomous Prefecture, Qinghai Province, China.
The concatenated seven replicase domains of Apodemus peninsulae CoV shared 98.9%–99.1% amino acid identities to the members of species in Coronavirus HKU24. Among them, the beta-CoV was detected from both Apodemus peninsulae and Microtus gregalis in Qinghai, China. The other members of the same species were previously detected from Rattus norvegicus in Guangdong (Lau et al. 2015), Apodemus agrarius in Zhejiang (Wang et al. 2015), Apodemus peninsulae in Tibet (Wu et al. 2018), Apodemus chevrieri in Yunnan (Ge et al. 2017), China and Rattus argentiventer in Vietnam (Phan et al. 2018). Those results indicate the members in Coronavirus HKU24 shared high nucleotide identity and may be commonly present in rats. However, the pathogenic potential remains unknown.
As a member of delta-CoV, PorCoV HKU15 was firstly reported in pigs without pathogenic evidence in Hong Kong of China in 2012, later detected in pig farms causing watery and acute diarrhea in U.S. in 2014, and then in Thailand, the Mainland of China, Korea and Lao PDR with 30%–40% death rate (Wang et al. 2014; Chen et al. 2015; Jung et al. 2015; Ma et al. 2015). PorCoV HKU15 shares the most recent common ancestor with SpCoV strains (HKU17, ISU42824, ISU690-4, ISU690-7 and ISU73347) suggesting the potential of cross-species transmission and/or shared host reservoir between pigs and sparrows. MtCoVs belong to the same CoV species with QuaCoV UAE-HKU30, PorCoV HKU15 and SpCoV HKU17, and all revealed phylogenetic positions shifting in phylogenetic trees based on different genes (Lau et al. 2018). And, species of hosts of Coronavirus HKU15 were diverse, such as quail (order Galliformes, family Phasianidae), sparrows (order Passeriformes, family Ploceidae) and Montifringilla taczanowskii (order Passeriformes, family Passeridae). For MtCoV, almost the entire S gene may be a recombinant from another more distant related delta-CoV. A part of S gene of MtCoVs shared above 70% nucleotide identity to ThCoV-HKU12 (Fig. 4B). The estimated tMRCA of MtCoV HM and ThCoV-HKU12 was around 600 years ago. So, the low nucleotide identity values of MtCoV HM and ThCoV-HKU12 in S gene (full length) indicated that the virus may has evolved to accommodate the new host after recombination (Lau et al. 2018). However, it is worth further investigating their cross-species transmission potential and mechanism.
Interestingly, we found two mostly identical sequences between MtCoV HM and MtCoV N. Firstly, the faecal samples of Montifringilla taczanowskii and Marmota himalayana were collected by Yushu Prefecture Center for Disease Control and Prevention at different times and different sites. Therefore, the cross-contamination of species was ruled out. Secondly, viral RNAs of faecal samples of Montifringilla taczanowskii and Marmota himalayana were extracted on different dates and sequenced as different pools. Thus, cross-contamination of the samples was also ruled out. Montifringilla taczanowskii and Marmota himalayana are common wild animals in Qinghai-Tibet plateau, and Montifringilla taczanowskii can enter marmot and pika caves (Ge et al. 2020). Because only one positive stool samples of Marmota himalayana was detected, the delta-CoV may be present accidentally in intestinal tract of the Marmota himalayana. The mechanism how Marmota himalayana got the delta-CoV still needs further study.
Overall, our study identified a novel delta-CoV and for the first time, found that Montifringilla taczanowskii may be a novel host of delta-CoV. This study facilitates a better understanding of the genetic diversity of delta-CoVs. Because the isolation was unsuccessful, the pathogenic potential of MtCoV is still unknown and requires further studies.
-
This work was supported by grants from National Science and Technology Major Project of China (2018ZX10712001-018, 2017ZX10303405-002, 2017ZX10303405-005-002), National Science and Technology of China (2017FY101202), National Key R & D Program of China (2019YFC1200500 and 2019YFC1200505), Sanming Project of Medicine in Shenzhen (SZSM201811071) and Research Units of Discovery of Unknown Bacteria and Function (2018RU010). The authors thank professor Changyun Ye for sharing the samples of rats.
-
JX and WZ conceived this study. WZ, JY, SL, XL, and SW collected the samples. WZ and DJ conducted experiments. WZ, JP and JY performed sequencing and analyzed the data. WZ, JY, and RL drafted the manuscript. JX finalized and supervised the study. All authors read and approved the final version of the manuscript.
-
The authors declare that they have no conflict of interest.
-
The study practices were approved by Ethical Committee of the National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention (NO: ICDC-2016004).

 DownLoad:
DownLoad: