












HTML
-
Since the discovery of the human immunodeficiency virus/ acquired immune deficiency syndrome (HIV/AIDS) pandemic ~40 years ago, it has become one of the largest global health problems. For example, 37.9 million individuals lived with HIV in 2019 (WHO 2020) and an additional 1.7 million became newly infected within the same year. There are two major types of the HIV, i.e. HIV- 1 and HIV-2 that both result in AIDS. Although HIV-2 is less pathogenic than HIV-1, both types share several similarities including basic gene arrangements, modes of transmission, intracellular replication pathways and clinical consequences (Nyamweya et al. 2013). The successful rollout of combination antiretroviral treatment (cART) strategies managed to prolong the life expectancy of HIV-1- infected individuals, shifting the disease to a chronic and manageable condition instead of an acute disease due to immunodeficiency (Nakagawa et al. 2013; Sabin 2013). Although most cART-treated HIV-1-infected individuals achieve viral suppression, this may not necessarily result in complete immunological recovery (Prabhakar et al. 2011; Pinzone et al. 2012; Wilson and Sereti 2013). The emphasis of this review article is therefore on HIV-1 as there is robust evidence that immune activation is the major driver of HIV-1-mediated pathogenesis (Lawn et al. 2001; Pandrea et al. 2008).
The early responses by innate immune cells to HIV-1 are important and may result in profound changes in immune signaling and the downstream production of cytokines and chemokines. Here the activation of innate immune cells leads to the induction of inflammation and the creation of an anti-viral type of status. Together such processes and related immune dysfunction can contribute to the establishment of immune activation and dysregulation. However, despite the importance of the innate immune system, most studies within the HIV field emphasize the role of the adaptive immune system and related pathological sequelae. In light of this, the current review focuses on the role of innate immunity (early to late changes) and its contribution to persistent immune activation and chronic immune dysregulation in HIV-1 infected individuals, with the spotlight on monocytes and macrophages. Such changes are also linked to downstream targets such as increased coagulation and the onset of cardiovascular diseases (CVD). It is our opinion that a detailed understanding of early mechanisms driving innate immunity together with the host immune cell response to HIV-1 is crucial to prevent HIV-mediated pathogenicity and to also help identify unique therapeutic targets.
-
Monocytes and macrophages are mediators of the innate immune system. Blood monocytes make up ~ 5% to 10% of total leukocytes and can circulate for up to three days before migrating to specific tissues. Here they can function as precursor cells to macrophages and (in some cases) to dendritic cells (DCs) (Yang et al. 2014). Recent findings also demonstrate that long-lived tissue resident macrophage populations (derived from yolk sac-progenitors and fetal liver-derived monocytes) are capable of self-renewal that can occur independently of circulating monocytes (Röszer 2018).
Mononuclear phagocytes are important antigen presenting cells, especially DCs and some subsets of monocytes, and to a lesser extent macrophages. These cells link the innate and adaptive immune systems, and hence play a central role in HIV-related pathogenesis (Anzinger et al. 2014; Merino et al. 2017; Kruize and Kootstra 2019). During the acute HIV-1 infection stage, early interactions between the virus and the immune system can impact on the viral load set-point and other serious events. For example, monocyte abnormalities are responsible for the immune hyperactivity observed in HIV-1-infected patients (Anzinger et al. 2014). Monocytes can respond to various stimuli, e.g. cytokines and microbial lipopolysaccharides (LPS) (Rossol et al. 2011) and therefore act as a crucial link between gastrointestinal translocation and immune activation during HIV-1 infection. Monocytes also trigger pro-inflammatory pathways that produce cytokines to initiate local and systemic inflammation (Schutte et al. 2009). In support, monocytes from HIV-infected persons with virological suppression produced relatively high levels of intracellular pro-inflammatory cytokines such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α compared to uninfected controls (Kedzierska and Crowe 2001; Kedzierska et al. 2003). It is also proposed that monocytes are responsible for chronic inflammation and co-morbidities (by regulation of inflammatory responses) in cART-treated HIV-1-infected individuals (Anzinger et al. 2014).
Monocytes are usually differentiated according to cell surface expression of phenotypic markers, cytokine production and gene expression profiles (Wong et al. 2011; Gren et al. 2015; Cormican and Griffin 2020). They are a heterogeneous population and each subpopulation possesses specific functions that mediate host defenses and inflammation. These cells can differentiate into classical (CD14+- CD16-) and CD16+ expressing monocytes (CD14+CD16+). The classical subtype constitute ~ 80% to 90% of total circulating monocytes while the CD14+CD16+ comprise the remainder. The CD14+CD16+ population is further subdivided into intermediate (CD14++CD16+) and non-classical monocytes (CD14+CD16++) (Ziegler-Heitbrock et al. 2010; Boyette et al. 2017). For classical monocytes the "classical" terminology is employed as their phenotype matches the original description of a monocyte (Ziegler-Heitbrock et al. 2010; Wong et al. 2011).Classical monocytes are described as CD14++ CD16- due to high CD14 expression (a component of the LPS receptor complex) together with no expression of CD16, a low-affinity receptor for human immunoglobulin G. Their main function is to facilitate phagocytosis, a process that is linked to reactive oxygen species generation (Ziegler-Heitbrock et al. 2010; Wong et al. 2011). They also produce cytokines such as TNFα and IL-1β, while expressinga relative high level of C–C chemokine receptor type 2 (CCR2) as homing markers.
The characteristics of intermediate monocytes—despite its name—do not simply fall midway between classical and non-classical monocytes. Intermediate monocytes comprise ~ 2% to 8% of circulating monocytes (Wong et al. 2011) and their functions include reactive oxygen species production, antigen presentation, participating in the proliferation and stimulation of T cells, inflammatory responses and angiogenesis (Wong et al. 2011; Boyette et al. 2017; Cormican and Griffin 2020). This subpopulation also expresses CCR2 at relatively low levels compared to classical monocytes, while expressing CCR5 as the main HIV receptor (Table 1). Such monocytes can migrate to the site of infection and invade tissues via CCR2/CC-chemokine ligand 2 (CCL2) (Si et al. 2010). Intermediate monocytes is a specialized subpopulation with the ability to secrete significant amounts of inflammatory cytokines, thus possessing robust pro-inflammatory potential and a prominent involvement in human atherosclerosis (Zawada et al. 2012).
Human monocyte Classical Intermediate Non-classical Surface markers CD14++CD16- CD14++CD16+ CD14+CD16++ Percentage (normal range) 80%–95% 2%–11% 2%–8% Chemokine receptor CCR2high CX3CR1low
CD62L+CCR2mid CX3CR1high
CCR5+CCR2low
CX3CR1highFunction Phagocytosis and immune response
Linked to normal immune responsePro-inflammatory
Linked to CVD, chronic inflammatory disease and HIVPatrolling and anti inflammatory
Inflammation and HIV infectionResemble mouse subsets Ly6C+ Ly6C+ Ly6C- Adhesion molecules CD62L expression High Low Low Response to LPS High IL-10 & low TNF-α levels IL-1β and TNF-α Low IL-10 and high TNF-α levels Gene signature Induction of T cell proliferation Links to T cell activation and antigen presentation Correlates with T cell activation Viral load (HIV) Decrease with HIV-1 infection Positive correlation Positive correlation CD4 Positive correlation Negative correlation Negative correlation TF/HIV infection Low expression Increased expression Increased expression D-dimer/HIV ND Positive correlation Positive correlation Data compiled from: Funderburg et al.(2010 , 2012);Wong et al. (2011) ; Woollard and Geissmann (2010);Yang et al. (2014) ; and Ziegler-Heitbrock(2007, 2015).
CCR: Chemokine receptor; CX3CR1: CX3C chemokine receptor 1; ND: Not determined; HIV: Human immunodeficiency virus; LPS: Lipopolysaccharides; Ly6: Lymphocyte antigen 6 C; TF: Tissue factor; TNF-α: Tumor necrosis factor-alpha.Table 1. Comparative analysis of the three described monocyte cell subpopulations.
Non-classical monocytes comprise ~ 2% to 11% of circulating monocytes (Ziegler-Heitbrock et al. 2010; Wong et al. 2011), are mobile in nature and patrol the endothelium in terms of injury. They express the CX3C chemokine receptor 1 (CX3CR1) at relatively high levels and can protect vessel walls against CX3CR1/CCL3 invasion in response to triggering stimuli (Geissmann et al. 2003). Moreover, non-classical monocytes with the Ly6C- phenotype are classified by some as being anti-inflammatory in nature as they play an important role to patrol the vascular endothelium (Buscher et al. 2017).
-
Immune activation during HIV-1 infection is a general phenomenon that includes several active molecular and intracellular processes such as cell activation, secretion of soluble molecules, proliferation and death, and their effects. HIV-1-infected individuals display elevated markers of immune activation (CD38 and human leukocyte antigen-DR [HLA-DR]) on CD8+ and CD4+ T cells which add to the prognostic value of low CD4+ T cell levels (Giorgi et al. 1993). Inflammation is an effector response of innate immune stimulation, and innate immune cells comprise phagocytes (granulocytes/monocytes/macrophages and DCs) as well as key lymphocyte populations such as natural killer (NK) and other innate lymphoid cells. Non-immune cells can also contribute to inflammation induction, including barrier cells (epithelial) of innate immunity. The inflammatory process is initiated by infection or injury when specialized pattern recognition receptors (PRRs) or damageassociated molecular patterns recognize pathogens. There are four major PRR sub-families i.e. toll-like receptors (TLRs), nucleotide-binding oligomerization domain-leucine rich repeats-containing receptors, retinoic acid-inducible gene 1-like receptors (RLR; aka RIG-1-like helicases— RLH), and C-type lectin receptors (Loo et al. 2008; Sabbah et al. 2009; Kumar et al. 2009). The engagement of PRRs on the innate immune cells promotes co-stimulatory signals for the adaptive immune cells (mainly T lymphocytes) (Land 2015). In addition, they activate microbicidal and proinflammatory responses essential to eradicate infectious agents, including the induction of cell death (Land 2015). Nucleotide-binding oligomerization domain-like receptors and RLRs are anti-viral members of the PRRs family that recognizes bacteria, viruses, fungi and protozoa, while NOD-like receptors (NLRs) can detect bacteria. It is likely that the interplay between such families supports the effective coordination of innate immune responses, e.g. important interactions between TLRs and certain NOD-like receptors can induce the production of pro-inflammatory cytokines (IL-1β) (Creagh and O'Neill 2006).
-
Acute inflammation starts immediately after HIV infection and is characterized by viremia. The latter can be confirmed by laboratory tests that include assessments of HIVRNA and p24 antigen detection (Palmer et al. 2003). This phase usually lasts up to 6 months and ends when anti-HIV antibodies are detected in plasma (Cohen et al. 2011). During acute infection there is a transient loss of circulating CD4 cells (Brenchley et al. 2004) while in parallel there is a significant depletion of CD4+ T cells (especially T-helper 17 [Th17] and effector memory Th cells) from the gastric mucosa due to disruption of lymphoid tissues and changes that occur in mucosal tissues (Brenchley et al. 2004). The early stages of infection result in damage to the gut mucosal epithelium and subsequent microbial translocation and systemic immune activation with chronic inflammation. The significant CD4+ T cell depletion from gut mucosa during the early stages of infection results in damage to the gut mucosal epithelium and subsequent microbial translocation. Here CCR5+ memory T cells with an activated phenotype are primarily affected. Cytokinemediated damage to gap junctions in the gut epithelium that is characterized by significantly higher production of TNF-α, IL-6, IL-8 and monocyte chemoattractant protein-1 is evident following HIV-1 exposure (Nazli et al. 2010; Brenchley 2013). Moreover, ongoing microbial translocation is linked to systemic immune activation and permanent changes to host immune cells (Brenchley et al. 2006; Cassol et al. 2010). Microbial translocation can in turn induce persistent monocyte and macrophage activation that is unrelated to the HIV viral load (Wallet et al. 2010).
Events occurring during acute HIV-1 infection are now recognized as crucial determinants of the subsequent disease progression (Borrow and Bhardwaj 2008). The innate immune system is the first line of defense and consists of immune cells that are able to rapidly recognize and respond to infections. The immune response to a viral infection starts when viral pathogen-associated molecular patterns are recognized by PRRs of infected cells that include TLRs. Meier et al. (2007) demonstrated that HIV-1 singlestranded RNA encodes for multiple TLR7/8 ligands that can mediate direct activation of the immune system (Meier et al. 2007). This subsequently triggers a signaling cascade that initiates innate intracellular anti-viral defences to restrict viral replication and its spreading. This response also produces cell-mediated and soluble factors including type Ⅰ and type Ⅲ interferon (IFN). Pro-inflammatory cytokines and chemokines can subsequently activate innate immune cells (including macrophages, NK cells and DCs) and attract them to the site of infection. Moreover, relatively high levels of pro-inflammatory cytokines such as TNFα, IL-6 and IL-1β are also detected (in plasma and lymph nodes) during the early stages of HIV-1 infection. There is also increased secretion of chemokines such as macrophage inflammatory protein-1α and 1β, and CCL5, in such patients (Sokol and Luster 2015). Anti-viral innate effector cells can later contribute to the control of viremia as well as the quality of the adaptive immune response to HIV-1 (Chang and Altfeld 2010; Carrington and Alter 2012).
What prevents the immune system from completely eradicating the HIV in this instance? There are several reasons that can help explain this phenomenon. In the first instance, HIV infects some CD4+ Th cells and also other cells that express CD4 (e.g. mononuclear phagocytes which includes DCs and Langerhans cells) and destroys a large number of CD4+ T cells, especially memory cells (Février et al. 2011). The loss of a key immune regulatory cell type will subsequently impact on other cells that make up the immune system. In addition, the invading virus can also establish latent reservoirs (not recognized by the immune system) in memory cells so that it can be reactivated at a later stage to once again replicate. Here recent discoveries show that monocytes, macrophages and vaginal epithelial DCs are potentially important long-lived HIV reservoirs (Pena-Cruz et al. 2018; Wong et al. 2019). Moreover, there is a suggestion that the shift from Th1 to Th2 cells and regulatory T cells may play a role in this instance (Klein et al. 1997; Christiaansen et al. 2015). However, this shift may also be due to the loss of Th1 cells. Another option is that the immune system tries to rectify the proinflammatory milieu by up-regulating "exhaustion" markers—such as programmed cell death protein markers and T cells immunoglobulin mucin domain-3—that in turn can render T cells non-functional (Boasso et al. 2009; Reuter et al. 2012). Finally, there may also be increased insensitivity of HIV to neutralized antibodies and cellular immunity as a result of its high mutation rate that allows it to escape immune surveillance and destructive systems (Mascola and Haynes 2013).
The link between immune activation and HIV pathogenesis is quite clear from studies of different monkey species infected with simian immunodeficiency virus (SIV). Here the natural host or non-pathogenic species (e.g. African Green Monkey or Sooty Mangabey) did not develop immune activation and AIDS despite a relatively high viral load (Pandrea et al. 2008). By contrast, pathogenic species (e.g. Rhesus macaque) developed AIDS with a relatively high level of immune activation (Pandrea et al. 2008; Klatt et al. 2012) (Table 2). These comparative analyses show that major differences are reflected by how host immune cells (innate immunity) respond to HIV (Christiaansen et al. 2015). For example, with nonpathogenic SIV infection the immune activation is dampened by increased IL-10 production, decreased T cell activation and attenuated apoptosis (Klatt et al. 2012). Moreover, levels of CD169—a marker of monocyte IFN-α activation—were also markedly increased in the Rhesus macaque, but decreased in the Sooty Mangabey (Jaroenpool et al. 2007; Pulliam 2014). This suggests that the Sooty Mangabey adapted to the SIV infection by changing their immune response and thereby escaped damage to their immune system due to its chronic activation (Jaroenpool et al. 2007; Pulliam 2014). Monocytes also became unresponsive to LPS stimulation and no longer secreted TNF-α, a powerful pro-inflammatory cytokine that impacts on the initiation of the immune response. Furthermore, there was an absence of microbial translocation in SIV natural hosts with a relatively low level of immune activation (Pandrea et al. 2008; Pulliam 2014). Thus it is apparent that the nature of the early response of innate immune cells plays a key role in terms of the degree of HIV-mediated pathogenesis (Mogensen et al. 2010).
Non-pathogenic species (e.g. African Green Monkey) Pathogenic species (e.g. Rhesus macaque) Stable viral replication High viral replication Normal level of immune activation and apoptosis High level of immune activation and apoptosis Restoration of CD4 T cells in peripheral blood and intestine No restoration of CD4 T cells Lower levels of CCR5 expressing CD4 T cells, with lack of disease progression Higher levels of CCR5 expressing CD4 T cells, with disease progression Preservation of immune cell subset function Abnormal immune cell subset function Lack of microbial translocation, with absence of aberrant immune activation High levels of microbial translocation, with aberrant immune cell activation Establishment of early anti-inflammatory milieu Lack of early anti-inflammatory role Early interferon response drops off Early interferon response continues CD169 expression decreases on monocytes CD169 expression increases on monocytes Reduced activation Chronic activation Table 2. Non-pathogenic vs. pathogenic models of simian immunodeficiency virus.
Together, the host innate immune cells response to HIV during the early stages of infection is an important contributor to persistent immune activation. The latter depends on how host innate immune cells respond to the virus, starting from the response of monocytes to LPS with microbial translocation, the secretion of pro- and antiinflammatory cytokines and the activation of different signaling pathways and its interactions. This can all contribute to continuous stimulation of the immune system and the manifestation of chronic inflammation in a systemic manner that can lead to the phenomenon of persistent immune activation that then occurs despite cART roll-out. Such immune activation can be detected by an expansion of CD38 and HLA-DR expressing CD8+ T cells (Kestens et al. 1994). In support, research shows that the coexpression of CD38 and HLA-DR decreases with cART but not to baseline levels as measured in non-infected persons (Al-Harthi et al. 2004). Together it is clear that HIV pathogenesis is driven by chronic inflammation that is strongly associated with infection and in concert with robust increases of inflammatory cytokines such as IL-6, IL-10, TNF-α, IFN-α and IFN-γ, and the link between cytokine induction and dysfunction during HIV pathogenesis (Reuter et al. 2012).
-
Residual levels of viral replication during cART are associated with persistent low levels of immune activation and inflammation, suggesting that unresolved inflammation can promote the replenishment of the HIV reservoir in tissues. In support, for the majority of virologically suppressed HIV-infected individuals on cART, reservoirs of latently infected cells (including resting CD4+ T cells and CD16+ monocytes) persist and contain replication-competent, latent provirus. This represents a major barrier to the eradication of HIV (Sonza et al. 2001; Zhu et al. 2002). Moreover, activated CD16+ monocytes are capable of disseminating HIV replication through ongoing cell-to-cell transfer of virions and effective infection of CD4+ T cells (Duncan et al. 2013).
TNF-α and mononuclear phagocytes (which includes monocytes, macrophages, DCs and Langerhans cells) are key drivers of HIV replication, disease progression and immune dysregulation. In agreement, there is a positive correlation between increased TNF-α and HIV-1-induced viremia as the former is one of the most powerful proinflammatory cytokines that can elicit severe inflammatoryrelated damage (Norris et al. 2006; Pasquereau et al. 2017). Here TNF-α and related receptors (TNFRs) likely play a key role in HIV-mediated pathogenesis, i.e. TNFR signaling controls HIV replication while HIV proteins interfere with TNF/TNFR pathways. TNF-α can also induce IL-6 and IL-8 that leads to the upregulation of viral replication (Ownby et al. 2009; Kumar et al. 2013). Moreover, IL-6 and TNF-α are necessary during acute stage reactions thereby contributing to fever and anorexia, processes characteristic of systemic inflammation. Hence the targeting of the TNF/TNFR pathway by novel therapeutic approaches may eventually assist in controlling immune activation and thus impact on both viral replication and persistence (Kumar et al. 2013; Pasquereau et al. 2017).
IFNγ plays various roles in terms of HIV-related pathogenesis. The production of IFNγ is detected early during the acute phase and continuously throughout the course of the infection. Although such production aims to clear the primary infection, it can (together with other inflammatory cytokines) induce chronic immune activation that intensifies clinical diseases. Unlike Type 1 IFNs, IFNγ has no direct antiviral activity against HIV-1 in primary cultures and can even enhance HIV-1 replication and associated diseases (Roff et al. 2014). By contrast, IFNγ can enhance cytotoxic T lymphocytes and NK cell activities against HIV-1 infected cells to help control HIV-1 replication (Roff et al. 2014).
In addition to such effects, modulators such as transforming growth factor beta and glycoprotein A repetitions predominant play a role in the transfer of Th1 to regulatory T cells with HIV infection, and this shift leads to an increasingly prominent role for Th2 and regulatory T cells as the disease progresses (Clerici and Shearer 1993; Miller et al. 2014; Christiaansen et al. 2015). However, the Th2 response is not enough to control HIV pathogenicity and this results in viral persistence and the development of chronic HIV-induced pathogenesis (Hunt 2007; Klatt et al. 2012; Paiardini and Müller-Trutwin 2013).
The induction of anti-inflammatory cytokines such as IL-10 decreases the inflammatory response by suppressing both inflammatory cytokine-mediated signaling and impaired DC and Langerhans cells maturation resulting from the inhibition of effector T cells (De Smedt et al. 1997). In contrast, IL-10 induction is crucial for supporting viral persistence and preventing viral clearance, e.g. upregulation of IL-10 and programmed cell death protein 1 by monocytes during HIV-1 infection leads to reversible CD4+ T cell dysfunction and impaired viral clearance (Said et al. 2010). Moreover, others found that the attenuation of IL-10 signaling induced viral clearance and resulted in the restoration of CD4+ and CD8+ T cell proliferation (Brooks et al. 2006; Wilson and Brooks 2011). This suggests that IL-10 may play a role in HIV-mediated immune exhaustion (Blackburn and Wherry 2007). As HIV infection is associated with increased IL-10 levels, this shifts Th1 to Th2 and regulatory T cells (Christiaansen et al. 2015) thereby leading to decreased activity of crucial immune cells responsible for viral clearance. This in turn leads to persistent viral infection and chronic inflammation that eventually results in increased morbidity and mortality.
Thus, although HIV promotes inflammation and immune activation which augments cytotoxic T lymphocytes, and assists with virus clearance, inflammation and immune activation also drives viral replication (Paiardini and Müller-Trutwin 2013). Conversely, IL-10 dampens inflammation and immune activation but also allows the invading virus to better replicate and perpetuate itself. It is important to note that a choice is required, i.e. either the virus is tolerated and reduced to relatively lower levels or it should be effectively eliminated when it becomes active as with an acute infection.
Early Stages and Role of Innate Immunity
Inflammation and HIV Replication: Role of Monocytes and Macrophages
-
Chronic HIV infection causes persistence of immune activation, upregulation of markers of inflammation, and apoptosis of host innate and adaptive immune cells (particularly monocytes, macrophages, NK and innate lymphoid cells). Although antiretroviral therapy restores CD4+ T cell counts, the persistent aberrant activation of monocytes and macrophages likely contributes to the incomplete recovery of T cell effector functions. This is further manifested by the effects of HIV infection and cART on the phenotype and function of circulating monocyte subsets and tissue macrophages. In addition to contributing to a latent reservoir, monocytes also play a role in terms of immune senescence and an increased risk of coagulation and CVD onset.
-
Latently infected CD4+ T cells comprise the majority of the HIV reservoir. Monocytes (mainly CD16+ monocytes) form an important part of this reservoir and also maintain HIV replication through ongoing cell-to-cell transfer of virions and infection of CD4+ T cells, while DCs and Langerhans cells play a significant role in transferring HIV to CD4+ T cells (Botting et al. 2017; Bertram et al. 2019). This can occur despite the availability and roll-out of cART. The latent reservoir consists of both CD4+ T cells and myeloid cells. CD4+ T cells and macrophages from various tissues can also contain replication-competent SIV and contribute to viral rebound due to interruption of treatment (Abreu et al. 2019). Additionally, circulating monocytes can contain latent viruses and contribute to the size of the latent reservoir after invading tissues and differentiating into long-lived macrophages (Abreu et al. 2019). This can occur despite circulating monocytes not usually being considered as a latent reservoir due to their relatively short life span.
-
LPS is a gram-negative bacterial product that is linked to immune activation with HIV infection (Brenchley et al. 2006). It directly activates mononuclear phagocytes which includes monocytes, macrophages, DCs and Langerhans cells. LPS stimulates monocytes/macrophages via the TLR4 that is associated with CD14 (Levy et al. 2009) and this leads to increased TNF-α secretion, the earliest and most potent pro-inflammatory cytokine. TNF-α displays contrasting roles in non-pathogenic versus pathogenic SIV infection. For non-pathogenic infections, immune activation is dampened by rendering monocytes insensitive to LPS and thereby decreasing TNF-α secretion while increasing IL-10 production (Klatt et al. 2012). IL-10 can in turn inhibit LPS–induced human monocyte tissue factor (TF) expression (Lindmark et al. 1998). During HIV infection there is a strong correlation between plasma LPS and immune activation, with the former contributing to immunodeficiency that can occur with chronic HIV infection (Brenchley et al. 2006). Moreover, LPS can also increase pro-coagulant TF expression on circulating blood monocytes, e.g. Funderburg et al. (2010) demonstrated a significant proportion of monocytes expressing TF in HIV-infected blood samples versus matched controls (Funderburg et al. 2010). These data indicate an important role for LPS in terms of monocyte TF induction that in turn can contribute to clotting and ultimately CVD onset in HIV-infected persons (Funderburg et al. 2012; Funderburg and Lederman 2014). This can be further compounded by monocyte TF expression preventing fibrinolysis and rendering clots resistant to heparin (Semeraro et al. 2009). The pivotal role of inflammation in terms of the development of thrombus formation has been the subject of many studies, supporting a correlation between inflammation and pro-thrombotic factors (Levi et al. 2003). For example, the SMART Study was the first to show a strong association between inflammation and coagulation, and CVD morbidity and mortality in HIV-infected individuals (Kuller et al. 2008). Others also demonstrated increased TF expression with HIV infection and that this correlated with viremia and T cell activation. This was the case especially for the activation of monocyte subsets where—for acute coronary syndrome patients—TF expression was robustly linked to non-classical monocyte subsets in HIV-positive patients (Funderburg et al. 2012). In support, our laboratory also demonstrated a strong link between TF (CD142) expression on activated T cells and non-classical monocyte activation in HIV-infected individuals (Teer et al. 2019).
-
Immune dysregulation during HIV infection can be directly linked to monocytes. Here the pathogenic sequence that occur in monocytes may contribute to systemic immune dysfunction characterized by excessive immune activation in infected individuals, which directly correlates with HIV pathogenesis and progression. Of note, some researchers investigated immune dysfunction in monocytes from untreated and treated HIV-positive patients and linked such findings to epigenetic changes (Espíndola et al. 2018). These data revealed that monocytes isolated from HIV patients exhibited dysfunctional phagocytosis together with dysregulated cytokine and reactive oxygen species production. Furthermore, they reported altered expression of enzymes regulating epigenetic changes during HIV infection and that this was more prominent in patients displaying relatively high levels of soluble CD163, a marker for macrophage activation and HIV progression (Espíndola et al. 2018). In agreement, our laboratory demonstrated a significant increase of CD163 in HIV-positive patients, especially for those exhibiting virological failure (Teer et al. 2019).
-
As discussed, persistent immune activation in HIV-infected individuals can lead to T cell activation. During a primary infection there is also an upregulation of telomerase, a crucial enzyme involved in the maintenance of telomere length. However, such upregulation is attenuated within the context of recurrent immune stimulation and hence memory T cells exhibit short telomere lengths that can lead to early or premature aging (Deeks 2011; Barsov 2011; Auld et al. 2016). Senescence occurs when the functional ability of immune cells is impacted by the permanent exhaustion of T cells (Chou and Effros 2013). The increasing turnover of new T cells then leads to premature aging of all the immune system cells and organs, with an accumulation of aging markers on T cells (CD28- and CD57+) (Brenchley et al. 2003; Chou and Effros 2013; Lee et al. 2014). Furthermore, peripheral blood monocytes from young HIVpositive individuals show changes in phenotype, function, and telomere length that closely resemble those detected in elderly controls aged ~ 30 years older (Deeks 2011; Hearps et al. 2012). Of note, such immune-related changes are not fully restored by cART.
With HIV infection, coinfection with cytomegalovirus (CMV) is associated with inflammation, immunological aging, and increased risk of severe non-AIDS related comorbidities. For example, Ballegaard et al. (2018) demonstrated that the magnitude of the CMV-specific T cell response was linked with a senescent immune phenotype. This may explain how a dysregulated immune response against CMV may contribute to immunological aging often described in people living with HIV, despite continuous and effective treatment (Ballegaard et al. 2018).
-
During HIV infection, monocytes display activated phenotypes that increase expression of the CD14+CD16+ population (Funderburg et al. 2012). The CD14+CD16+ monocyte subset is preferentially susceptible to HIV-1 infection as it expresses relatively high CCR5 levels (Ellery et al. 2007). The identification of such subpopulations during HIV infection is necessary to better understand monocyte function, especially as monocyte perturbations can still exist despite the availability of cART. For example, cART-treated individuals displayed an expansion of intermediate and non-classical monocytes (Teer et al. 2019). The expansion of both intermediate and nonclassical monocyte subsets is proposed to play an important role in HIV-mediated pathogenesis, especially CVD onset and progression (Funderburg et al. 2012). The significance of CD16+ monocytes in atherosclerosis is highlighted by its pro-inflammatory capacity and increased ability to produce cytokines (IL-6), matrix metalloproteinases, monocyte chemoattractant protein-1, as well as their potential role to activate endothelial cells (Belge et al. 2002; Ghattas et al. 2013). Classical monocytes play a crucial role during the onset of acute myocardial infarction and play a role in the early resolution of inflammation (Nahrendorf et al. 2007). By contrast, CD16+ monocytes are involved in the latter stages of inflammation and secrete relatively low levels of IL-10 (Nahrendorf et al. 2007).
Intermediate monocytes increases during HIV infection and is an independent predictor for CVD events and may be explained by relatively high levels of pro-inflammatory cytokines secreted with the expansion of this subset (Rogacev et al. 2012). Intermediate monocytes showed in vitro augmented lipid accumulation and oxidized low-density lipoprotein cholesterol uptake with lower cholesterol efflux compared to other subsets. Furthermore, intermediate monocytes revealed relatively high expression of cholesterol scavenging receptors (CD36, CD68) but lowered expression of the ATP-binding cassette transporter (implicated in cholesterol efflux) (Rogacev et al. 2014). Non-classical monocytes also display pro-inflammatory behavior and secrete inflammatory cytokines in response to infection, while also involved in antigen presentation and T cell stimulation (Mukherjee et al. 2015; Ong et al. 2018). Human non-classical monocytes are a heterogeneous population that comprise several subsets with functional differences (Hamers et al. 2019). Non-classical monocytes show high migratory but only limited phagocytic potential (Mukherjee et al. 2015) and such subsets exhibit altered frequencies in the setting of severe coronary artery disease (Hamers et al. 2019). The CD16+ monocyte population also increases significantly with infections, even when the latter is of a localized nature, and some reported a correlation between CD16 expression, TGF-b and elevated TNF-α serum levels (Belge et al. 2002).
CD14+CD16++ monocytes are also considered an inflammatory monocyte subset that strongly correlates with atherosclerosis and CVD (Hilgendorf and Swirski 2012; Stansfield and Ingram 2015). The CD16+ population is the preferential target for HIV infection as it expresses a relatively high level of CCR5 (Ellery et al. 2007). Recent research work also demonstrated the expansion of both non-classical and intermediate monocytes correlated with viremia and T cell activation in HIV-infected long-term non-progressors (elite controllers) (Prabhu et al. 2019). A study investigating the effects of 2 h of LPS treatment on TNF-α mRNA levels in the three monocyte subsets revealed that the non-classical subset showed a 150-fold upregulation of TNF-α, while the classical and intermediate subsets each showed only ~ 50-fold upregulation (Ong et al. 2018). This further demonstrates the robust proinflammatory role of non-classical monocytes that can be attributed to senescence (Ong et al. 2018). In support, some demonstrated that intermediate and non-classical monocytes showed relatively higher cytokine production (TNFα, IL-1β, IL-6) and M1 marker (CD86) expression, but lower levels of M2 markers (CD93, CD163) compared to classical monocytes (Patel et al. 2017) indicating that the former two subtypes are more inflammatory in nature. Some studies also showed that in vitro TLR stimulation causes non-classical monocytes to generate low pro-inflammatory stimuli, producing TNF-α and IL-6, and that this was the most pro-inflammatory response under such conditions (Ong et al. 2018). They also found an accumulation of the non-classical monocytes together with higher plasma TNF-α and IL-8 levels in elderly persons. These factors may contribute to inflammaging and agerelated inflammatory conditions (e.g. atherosclerosis, osteoarthritis), indicating that the non-classical monocyte subset is a senescent population (Ong et al. 2018). Here the non-classical subset displayed relatively high expression of senescence markers, followed by the intermediate and the classical subsets (Ong et al. 2018).
Data generated in our laboratory support such notions. For example, we found that there is expansion of proatherogenic monocyte subsets (intermediate and nonclassical monocytes) with HIV infection of a South African cohort (Teer et al. 2019). Here non-classical monocytes correlated positively with immune activation (CD38) and coagulation markers (CD142) expressed on CD4+ and CD8+ T cells, indicating an association with such pathogenic processes during HIV infection. Our data also revealed a positive correlation between monocyte subsets and regulatory T cell activation markers glycoprotein A repetitions predominant and special AT-rich sequence binding protein 1, indicating the dual anti- and pro-inflammatory role of monocyte subsets and reflecting the imbalance of proand anti-inflammatory states during HIV infection (Teer et al. 2019). Thus the role of each monocyte subset during HIV-1 infection is a crucial factor to consider especially within the context of inflammation, coagulation and CVD development (Table 1).
Latent Reservoir
Monocytes/Macrophages Activation and Coagulation
Monocyte Dysfunction and Association with Epigenetic Changes
Immune Senescence
Altered Monocyte Phenotypes and Risk for CVD Onset
-
Monocytes and macrophages are key components of the innate immune system and play crucial roles in HIV pathogenesis. This starts from the early response to HIV, the involvement in terms of viral replication, and by acting as a viral reservoir throughout the course of the infection. They are also involved in regulation and initiation of the chronic inflammation and play a key role in the onset and progression of immune activation-related pathological sequelae (refer Fig. 1). In light of this, we recommend that future therapeutic interventions should aim to target monocytes and macrophages to selectively modulate aberrant activation and dampen pathological events in HIV-infected persons.
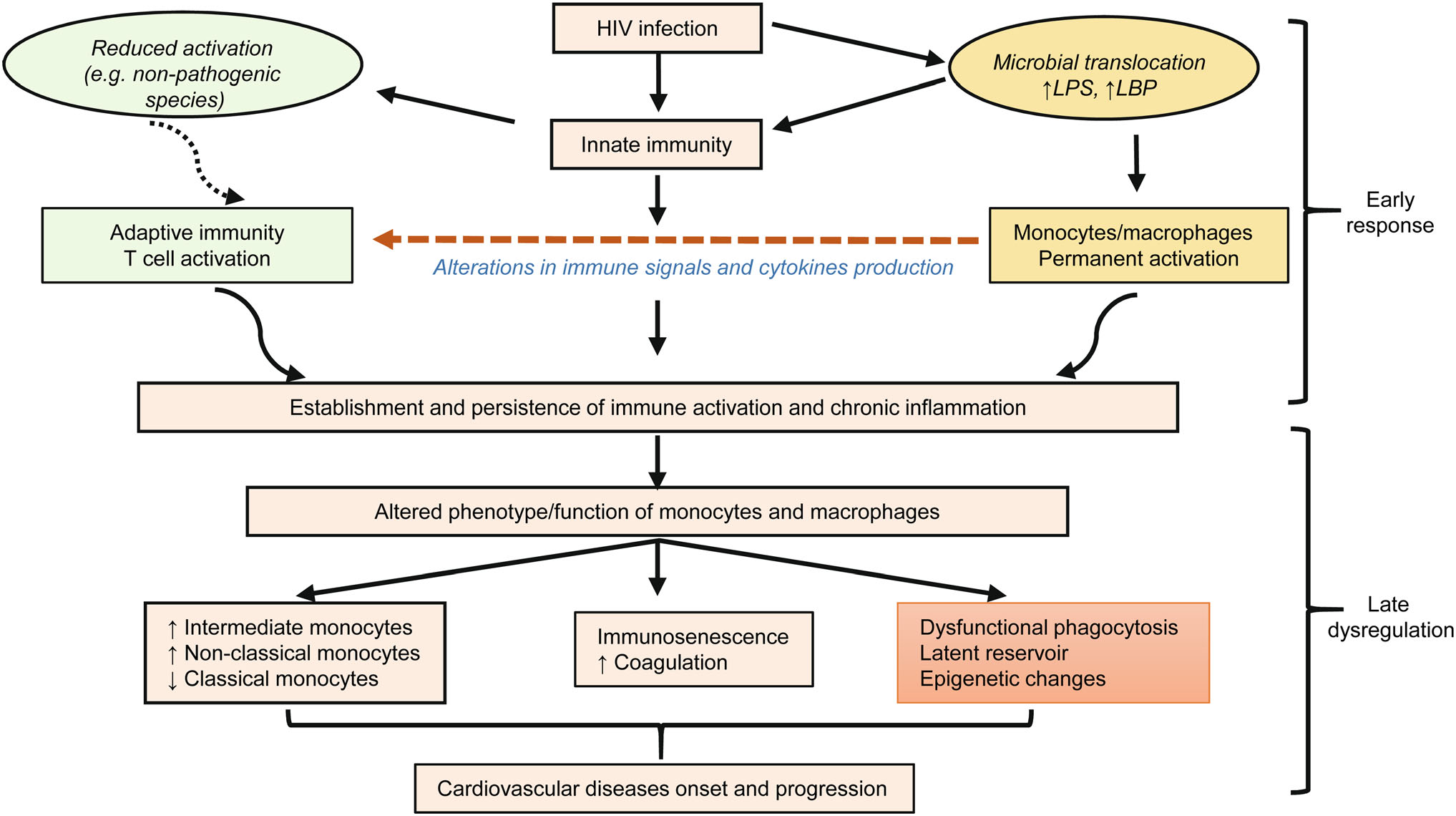
Figure 1. Role of monocytes and macrophages in HIV-1 infection from early response to late dysregulation. HIV infection leads to microbial translocation, subsequently inducing permanent activation of innate immune cells (monocytes and macrophages). This activation is reflected by the host innate immune cells' response to HIV infection during the early stages. Activation of monocytes and macrophages lead to the establishment and maintenance of immune activation, and chronic inflammation. This is further complicated with the persistence of the virus due to HIV reservoirs in monocytes and macrophages as well as abnormal phagocytic function. Furthermore, this lead to expansion and alteration of proinflammatory phenotype and function of monocyte subsets, also immuno-senescence as well as an increase in coagulation and CVD risk. HIV: Human immunodeficiency virus; LPS: lipopolysaccharide; LBP: lipopolysaccharide-binding protein.
-
The authors wish to acknowledge financial support provided by the South African Medical Research Council (to MFE).
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: