












HTML
-
Enteroviruses (EVs) including Enterovirus 71 (EV71), Coxsackievirus A16 (CVA16), CVA6, CVB3 and EV-D68 and so on, belong to the Enterovirus genus of the Picornaviridae family, which are positive, single-stranded RNA viruses. The viral genome is approximately 7500 nucleotides in length, with a single open reading frame that encodes a large precursor protein. Upon infection, the precursor protein is processed into four structural (VP1, VP2, VP3 and VP4) and seven nonstructural (2A, 2B, 2C, 3A, 3B, 3C and 3D) proteins (McMinn 2002). EVs infection is closely associated with hand, foot and mouth disease (HFMD), which has been identified as a class C infectious disease in the mainland of China since 2008 (Zhu et al. 2011). HFMD usually manifests most frequently as childhood exanthema, sometimes accompanied by severe neurological diseases including aseptic meningitis, brain stem or cerebellar encephalitis, as well as acute flaccid paralysis (McMinn 2002).
Innate immunity is the first line of defense against a viral invasion. In order to elicit type Ⅰ interferon (IFNs), pattern recognition receptors (PRRs) including RIG-I-like receptors (RLRs) were evolved by mammalian hosts to sense the presence of viral nucleic acids or other molecular components of invading microbes. However, viruses have developed various strategies to escape from host attack through downregulating the expression of RIG-I and MDA-5, as well as downstream effectors that ultimately generate IFN responses (Barral et al. 2007, 2009; Lei et al. 2013; Wang et al. 2013; Feng et al. 2014). TRIM25, containing an N-terminal RING domain with potential ubiquitin E3 ligase activity, represents a new class of antiviral molecules involved in innate immunity (Nisole et al. 2005; Ozato et al. 2008). TRIM25 has been shown to induce Lys 63-linked ubiquitination of the N-terminal CARDs of RIG-I, which is crucial for the cytosolic RIG-I signaling pathway to elicit host antiviral innate immunity (Gack et al. 2007). Specifically, TRIM25 interacts with the 1st CARD of RIG-I, and this interaction effectively delivers the Lys 63-linked ubiquitin moiety to the 2nd CARD of RIG-I, leading to efficient interaction with MAVS/VISA/IPS-1/Cardif (Xiao et al. 2011). Additionally, a splice variant of RIG-I which carries a deletion (amino acids 36–80) within the 1st CARD, loses TRIM25 binding, CARD ubiquitination and downstream signaling ability, demonstrating the critical role of TRIM25-mediated ubiquitination for RIG-I antiviral activity (Gack et al. 2008).
3C protease (3Cpro) of picornaviruses plays a pivotal role in viral replication and virus-host interactions. 3Cpro can induce apoptosis (Li et al. 2002, 2017; Croft et al. 2017), cleave certain cellular factors required for transcription, translation and activation of innate immunity (Kuyumcu-Martinez et al. 2004; de Breyne et al. 2008; Lei et al. 2011, 2013; Mukherjee et al. 2011). As strong antagonists of IFNs, EV-D68 and EV71 3Cpro downregulate or directly cleave interferon regulatory factor 7 (IRF7) and IRF9, the downstream molecule of TLRs and TLR signaling pathway, and inhibit innate antiviral immunity (Hung et al. 2011; Lei et al. 2013; Xiang et al. 2015). Coincident with these findings, EV71 3C was shown to block type Ⅰ IFN synthesis in a mouse model (Lee et al. 2012). Moreover, EV71 3Cpro can cleave IRF7 to block the JAK1-STAT signaling pathway (Hung et al. 2011). Increasing evidence suggests that EV71 3Cpro is required for viral evasion of innate immune responses under physiologic conditions (Lee et al. 2012; Hung et al. 2011; Wang et al. 2015; Lei et al. 2014). However, whether EVs affect RIG-I through regulating the expression of TRIM25 is undefined until now, and the underlying molecular mechanism needs to be further investigated.
In this study, we found that EV71, CVB3, CVA6 and EV-D68 but not CVA16 3Cpro could reduce not only RIG-I expression but also TRIM25 expression. Moreover, overexpression of TRIM25 could rescue the expression of RIG-I reduced by 3Cpro and restore the production of IFNs inhibited by 3Cpro. Further investigation showed that L69 and V72 amino acids in TRIM25 required for RIG-I ubiquitination could not rescue RIG-I expression and IFN production. Therefore, TRIM25-mediated RIG-I ubiquitination inhibited by the 3C protein may be the key contributor to the stability of RIG-I and suppression of type Ⅰ IFN production.
-
Human embryonic kidney 293T (HEK293T, CRL-11268) cells were purchased from ATCC and maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT, USA), 100 U/mL penicillin and 100 μg/mL streptomycin at 37 ℃ in a 5% CO2 humidified atmosphere. Sendai virus (SeV) was kindly provided by Junliang Chang (Jilin University, Jilin, China). EV71 (CC063) and CVA6 (changchun046/CHN/2013) viruses were isolated from HFMD patients by our laboratory. CVA16 (shzh05-1) and EV-D68 (US/KY/14–18, 953) viruses were gifted from Qi Jin (Institute of Pathogen Biology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing) and John Hopkins University, respectively.
-
To construct pRIG-I-Flag, RIG-I fragments tagged with Flag were cloned into the SalI and BamH I sites of the VR1012 vector. To construct pEV71-3C, 3C fragments of EV71 cDNA were cloned into the HindIII and SalI sites of the VR1012 vector. pEV71 P1-Myc was constructed by inserting the fragment into the SalI and Xba I sites of the VR1012 vector. Flag-RIG-IN, an RIG-I mutant containing the N-terminal domain (amino acids 1 to 242), was generously gifted by Jinhua Yang (Baylor College of Medicine, Houston, TX, USA). RIG-IN K172R mutant was made by site-directed mutagenesis. The EV71, CVB3, CVA16, CVA6 and EVD68 3C-HA were amplified from EV71 (GenBank #AF30299.1), CVA16 (Genbank #KF055238.1), CVA6 (Changchun046/CHN/2013 7434 KT779410), CVB3 (Genbank #AJ295194) and EV-D68 (AY426531.1) viruses and constructed by inserting the fragment into the NotI and HindIII sites of pcDNA3.1. The resulting constructs were confirmed by sequencing. The pGL3–IFN-β–Luc plasmid was constructed as descried previously (Rui et al. 2017). The TRIM25 plasmid was purchased from Addgene (Cambridge, MA, USA). The TRIM25 RING-Myc, TRIM25 B-box-Myc, TRIM25 CCD-Myc, TRIM25 SPRY-Myc, TRIM25-L69A and V72A-Myc mutant plasmids were cloned into the VR1012 vector. The EV71 3C-HA variants H40G, E71A and C147G were constructed by site-directed mutagenesis.
-
The following antibodies were used for Western blotting analysis in this study: anti-HA monoclonal antibody (MMS-101R, Covance, USA), anti-Myc monoclonal antibody (MMS-150P, Covance, Princeton, NJ, USA), anti-Tubulin (RM2003, Beijing Ray Antibody Biotech, Beijing, China), anti-Flag (F1804, Sigma, St. Louis, MO, USA), anti-RIG-I (AP1900A-400, Abgent, San Diego, CA, USA), anti-TRIM25 (H00007706-B01P, Abnova, Taiwan), anti-TRIM21 (12, 108-1-AP, Proteintech, USA), anti-PCBP2 (15, 070-1-AP, Proteintech, USA), anti-EV-D68 VP1 polyclonal antibody (GTX132313, GeneTex, USA), anti-CVA6 VP1 polyclonal antibody (GTX132346, GeneTex, USA). Polyclonal antibody (pAb) against EV71 and CVA16 viruses was obtained from rabbit immunized with EV71 or CVA16 whole viruses in our laboratory. The EV71 VP1 rabbit polyclonal antibody was produced in our lab.
-
HEK293T cells were plated into 24-well dishes and transfected the following day. One hundred nanograms of the reporter plasmid for IFN-β promoters, 1 ng Renilla luciferase control plasmid (pRL-TK) and the indicated amounts of the expression plasmids were used per well. At 24 h post-transfection, cells were infected with SeV (20 HA/mL) or left uninfected for 18 h. Luciferase activities were then measured using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer's instructions. Firefly luciferase activity was normalized to Renilla luciferase activity. The relative luciferase activities (Rel. Lucif. Act) were expressed as fold changes over that of the empty plasmid transfected cells or SeV non-infected controls.
-
Total RNA was extracted from cells by using Trizol reagent (Invitrogen, Carlsbad, CA, USA). RNA samples were treated with DNase I (Promega, Madison, WI, USA), and reverse transcription was carried out using a Superscript cDNA synthesis kit (Invitrogen, Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. cDNA samples were subjected to PCR amplification and electrophoresis to detect IFN-β, ISG15 and ISG56 expression. Primers used were as follows: human IFN-β, AAACTCATGAGCAGTCTGCA and AGGAGATCTTCAGTTTCGGAGG; human ISG15, TCCTGGTGAGGAATAACAAGGG and CTCAGCCAGAACAGGTCGTC; human ISG56, TCGGAGAAAGGCATTAGATC and GACCTTGTCTCACAGAGTTC; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), TGCACCACCAACTGCTTAGC and GGCATGGACTGTGGTCATGAG. To quantify IFN-β gene expression, SYBR green-based qRT-PCR was carried out using a CFX96 system (Roche, Basel, CH, Germany). Expression of IFN-β was normalized to GAPDH mRNA expression. Primers used for IFN-β and GAPDH were the same as indicated above.
-
Cells were harvested and lysed in RIPA lysis buffer (50 mmol/L Tris–HCl, pH7.5, 150 mmol/L NaCl, 1% NP40), and lysates were cleared by centrifugation at 16, 000 ×g for 5 min at 4 ℃. Total cell extracts were subject to SDS-PAGE and transferred to nitrocellulose membranes (10, 401, 196; Whatman, Maidstone, UK). After blocking with 5% nonfat dry milk in TBST for 1 h at room temperature (RT), membranes were incubated with the indicated primary antibodies at 4 ℃ overnight and then the corresponding alkaline phosphatase (AP)-conjugated secondary antibodies (Sigma) for 1 h at RT. After three washes with TBST, the blots were reacted with nitroblue tetrazolium (NBT) and 5-bromo-4-chloro-3-indolylphosphate (BCIP) (Sigma). After three washes with TBST, the membranes were reacted with an ECL sensitive kit (B500023; Proteintech, Rosemont, IL, USA) and developed by using an Azure c500 imaging system (Azure Biosystems, Dublin, CA, USA).
-
At 48 h after transfection, cells were harvested and lysed with buffer (50 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 0.5% NP-40) containing a protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Lysates of cells were incubated with anti-Myc affinity matrix (Sigma A-2220) or anti-HA affinity matrix (Roche) at 4 ℃ overnight on a rotator. After rinsing with wash buffer (20 mmol/L Tris-Cl, pH7.5, 100 mmol/L NaCl, 0.05% Tween-20, 0.1 mmol/L EDTA) six times, 50 μL of elution buffer (100 mmol/L Glycine-HCl, pH 2.5) was added to re-suspend the beads, and the eluted proteins were obtained by centrifugation, followed by SDS-PAGE and Western blot analysis.
-
The detailed statistical analysis has been described in figure legends. All data are expressed as the mean ± standard deviations (SDs). Statistical comparisons between two groups were made using a Student's t-test. Significant differences are indicated in figures as follows: *P < 0.05, **P < 0.01 and ***P < 0.001. P-values of less than 0.05 are considered to represent a statistically significant difference. ns stands for no significance.
Cell Lines, Viruses
Plasmids
Antibodies and Reagents
Luciferase Reporter Assays
RNA Extraction and Quantitative Real-Time PCR (qRT-PCR)
Western Blot Analysis
Coimmunoprecipitation
Statistical Analysis
-
Previous studies have demonstrated that 3C proteins of EV71, CVA16 and CVB3 could inhibit RIG-I-mediated type Ⅰ IFN responses (Lei et al. 2010; Rui et al. 2017). In order to investigate how 3C proteins affect the innate immune pathway, we first detected the effect of EV71 and CVB3 3C proteins on RIG-I expression. RIG-I plus VR1012 or increasing amounts of EV71 or CVB3 3C expression vector were transfected into HEK293T cells as indicated. At the same time, the IFN-β luciferase reporter (Rui et al. 2017) and an internal control pRL-TK renilla reporter were transfected into HEK293T cells and used to measure the IFN response. 3C proteins of EV71 and CVB3 both reduced the expression of RIG-I in a dose-dependent manner (Fig. 1A and 1D). As reported, they both inhibited IFN-β promoter activity and downstream genes of IFN-β, ISG15 and ISG56 mRNA in a dose-dependent manner (Fig. 1B, 1C and 1E, 1F). The CARD domain located at the N-terminal of RIG-I is responsible for transducing signals to the downstream adaptor MAVS through CARD-CARD interactions (Rui et al. 2017). A RIG-IN expression vector, which includes the caspase recruitment domain, was employed to detect whether the CARD domain is the target of 3C. As illustrated in Fig. 1G and 1J, 3C proteins of EV71 and CVB3 could significantly reduce the expression of RIG-IN, as well as suppress RIG-IN-mediated IFN-β activation in a dose-dependent manner (Fig. 1H, 1I and 1K, 1L).
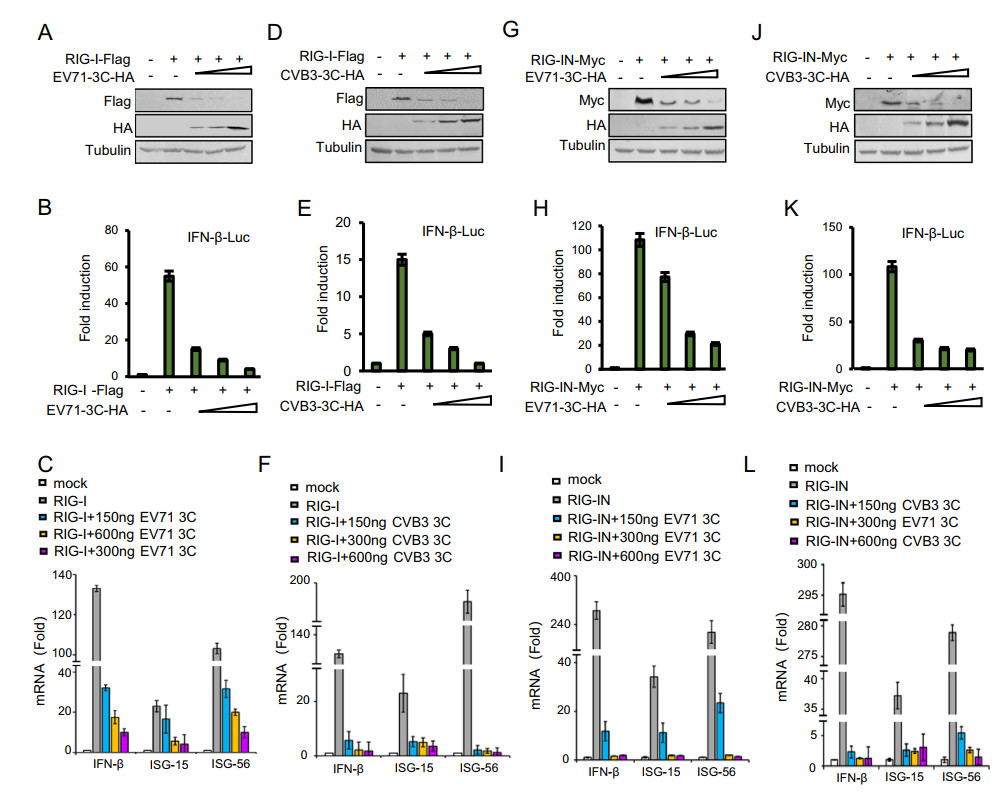
Figure 1. The 3C proteins of EV71 and CVB3 reduce RIG-I expression and inhibit IFN-β promoter activation. A, D, G and J Cell extracts prepared from HEK293T cells cotransfected with the IFN-β-Luc firefly luciferase reporter plasmid, the Renilla luciferase control reporter plasmid pRL and RIG-I or RIG-IN (300 ng) in the absence or presence of increasing dose (150 ng, 300 ng, 600 ng) of 3C plasmids as indicated for 48 h were subjected to immunoblotting with anti-HA, anti-Flag and anti-Tubulin antibodies. B, E, H and K Luciferase activities of cell lysates were analyzed with the Dual-Luciferase Reporter Assay System and measured on a Monolight 2010 luminometer. C, F, I and L Total RNA was extracted from HEK293T cells expressing the indicated plasmids. The IFN-β, ISG15 and ISG56 mRNA expression levels were determined by qRT-PCR. Data represent the average of three independent experiments. The error bars indicate the standard deviation (SD) of three replicates within one experiment.
We also examined whether endogenous RIG-I was reduced by 3C proteins of EV71 and CVB3 by applying an anti-RIG-I antibody after SeV infection in order to exclude non-specific effects. The results showed that SeV-induced RIG-I expression was also reduced by 3C proteins (Fig. 2A and 2B). TRIM25 has been reported to be critical for RIG-I ubiquitination and RIG-I antiviral activity. At the same time, we also observed that the 3C proteins reduced the expression levels of endogenous TRIM25 induced by SeV in a dose-dependent manner (Fig. 2A and 2B). Previous studies had reported that 3C of members of the family Picornaviridae can inhibit host cell translation early in infection (Etchison et al. 1982; de Breyne et al. 2008), so we detected whether EV71 3C affects the expression of other host proteins such as TRIM21 and PCBP2. The results showed that the expression of another TRIM family member TRIM21 and PCBP2 was not reduced by EV71 3C (Fig. 2C, 2D), indicating that the effect of 3C on RIG-I and TRIM25 expression is relative specific. Taken together, these results showed that the expression levels of RIG-I and TRIM25 both were significantly suppressed by 3C proteins of EV71 and CVB3 in a dose-dependent manner.
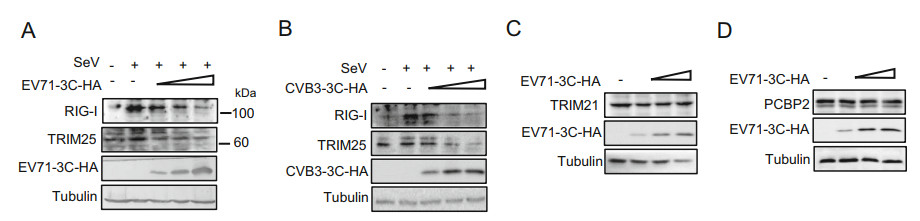
Figure 2. The 3C proteins of EV71 and CVB3 specifically reduce expression levels of the SeV-induced endogenous RIG-I and TRIM25 proteins. HEK293T cells were transfected with increasing amounts of the EV71-3C-HA plasmid (A) or CVB3-3C-HA plasmid (B) for 24 h. The cells were subsequently infected with SeV (20 hemagglutination [HA] units/mL) for 24 h, and then the cell lysates were subjected to immunoblotting with anti-HA, anti-RIG-I or anti-TRIM25 antibodies. C and D EV71 3C had no effect on the expression of TRIM21 and PCBP2. Data are representative of three independent experiments.
-
TRIM25-mediated ubiquitination can stabilize the RIG-I 2CARD tetramer and play a fundamental role in RIG-I-mediated antiviral activity (Gack et al. 2007; Sanchez et al. 2016). To further examine whether reduced expression of RIG-IN was restored by overexpressing TRIM25, RIG-IN-Myc and EV71 (Fig. 3A) or CVB3 (Fig. 3D) 3C-HA expression vector were transfected into HEK293T cells with increasing amounts of TRIM25-Myc. The IFN-β luciferase reporter and an internal control pRL-TK renilla reporter were also transfected into HEK293T cells and used to measure the IFN response as indicated. At 48 h post-transfection, cells were harvested for Western blotting and IFN-β promoter activity analysis. We found that RIG-IN expression was markedly increased by overexpressing TRIM25 protein in a dose-dependent manner (Fig. 3A and 3D). Moreover, reduced IFN-β promoter activity due to 3C proteins was significantly reactivated (Fig. 3B and 3E), and IFN-β mRNA production was partially upregulated (Fig. 3C and 3F). These results showed that TRIM25 could stabilize RIG-I by inhibiting 3C-mediated degradation of RIG-I, thereby inducing IFN signaling activation.
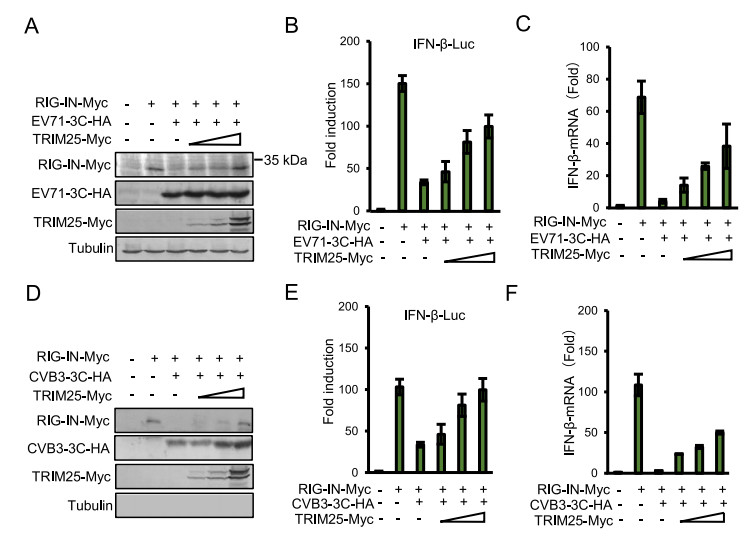
Figure 3. TRIM25 rescue IFN-β expression through mediating RIG-I ubiquitination. A and D HEK293T cells were co-transfected with the IFN-β-Luc firefly luciferase reporter plasmid, the Renilla luciferase control reporter plasmid pRL and 300 ng of RIG-IN, 300 ng of EV71/CVB3 3C, increasing dose (50 ng, 150 ng, 450 ng) of TRIM25 plasmids as indicated for 48 h, and then the cells lysates were subjected to immunoblotting with anti-Myc, anti-HA or anti-Tubulin antibodies. B and E Luciferase activities of the cell lysates were analyzed with the Dual-Luciferase Reporter Assay System and were measured on a Monolight 2010 luminometer. C and F Total RNA was extracted from HEK293T cells expressing the indicated plasmids, and then the IFN-β mRNA expression levels were determined by qRT-PCR. Data represent the average of three independent experiments.
-
TRIM25 contains a cluster of a RING-finger domain, a B-box domain, a coiled-coil domain and a SPRY domain (Fig. 4A). The two mutants L69A and V72A located in the RING domain of TRIM25 were shown to disrupt RING dimerization, which is required for RIG-I ubiquitination (Sanchez et al. 2016). In order to dissect whether the TRIM25-mediated rescue of RIG-I is closely associated with its ubiquitination function, we first detected whether TRIM25 L69A and V72A mutants could rescue the reduction of RIG-I expression mediated by 3C. The results showed that the L69A and V72A mutants could not restore the expression of RIG-I reduced by 3C proteins (Fig. 4B and 4C), suggesting that TRIM25-mediated ubiquitination of RIG-I is critical for RIG-I stability. Consistent with the expression of RIG-I, reduced IFN-β promoter activity due to 3C protein could not be restored by the TRIM25 L69A and V72A mutants (Fig. 4D and 4E). Moreover, we observed that EV71 3C protein didn't reduce the expression of RIG-IN K172R mutant, which is the ubiquitination site in RIG-I required for TRIM25 (Fig. 4F), indicating that RIG-I ubiquitination is necessary for 3C-mediated downregulation. In order to determine which domain of TRIM25 has a similar ability as wild-type (WT) TRIM25 to restore the expression of RIG-I and rescue RIG-I-mediated IFN-β stimulation, we constructed four TRIM25 truncated mutants. Surprisingly, the RING, B-box, CCD or SPRY domain of TRIM25 alone could not restore the 3C-mediated reduction of RIG-I expression and inhibition of IFN-β response (Fig. 5). These results suggest that the structural conformation of TRIM25 maintained by various domains essential for RIG-I ubiquitination also is required for RIG-I recovery.
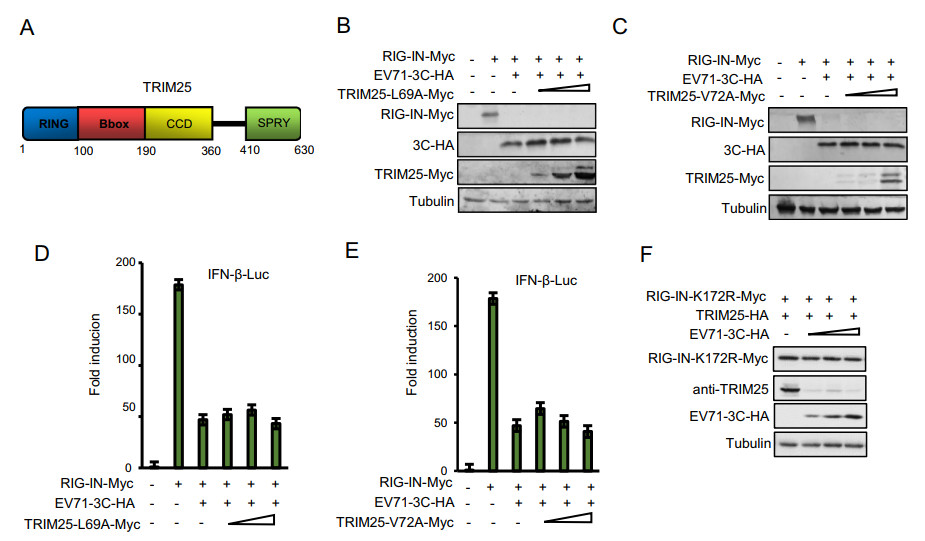
Figure 4. L69 and V72 amino acids in TRIM25 required for RIG-I ubiquitination can not rescue RIG-I expression and release the inhibition on IFN-β activation mediated by 3C proteins. A TRIM25 structural diagram. HEK293T cells were co-transfected with the IFN-β-Luc firefly luciferase reporter plasmid, the Renilla luciferase control reporter plasmid pRL, RIG-IN-Myc, EV71-3C-HA and increasing amounts of TRIM25-L69A-Myc (B) or TRIM25-V72A-Myc (D) for 48 h, and then the cells lysates were subjected to immunoblotting with anti-Myc, anti-HA or anti-Tubulin antibodies. C and E Luciferase activities of the cell lysates were analyzed with the Dual-Luciferase Reporter Assay System and measured on a Monolight 2010 luminometer. Data represent the average of three independent experiments. F Ubiquitination site K172 in RIG-I was not affected by EV71 3C expression. HEK293T cells were co-transfected with RIG-IN-K172R mutant, TRIM25-HA and negative control vector or increasing amounts of EV71-3C-HA for 48 h, and then the cells lysates were subjected to immunoblotting with anti-Myc, anti-TRIM25, anti-HA or anti-Tubulin antibodies.
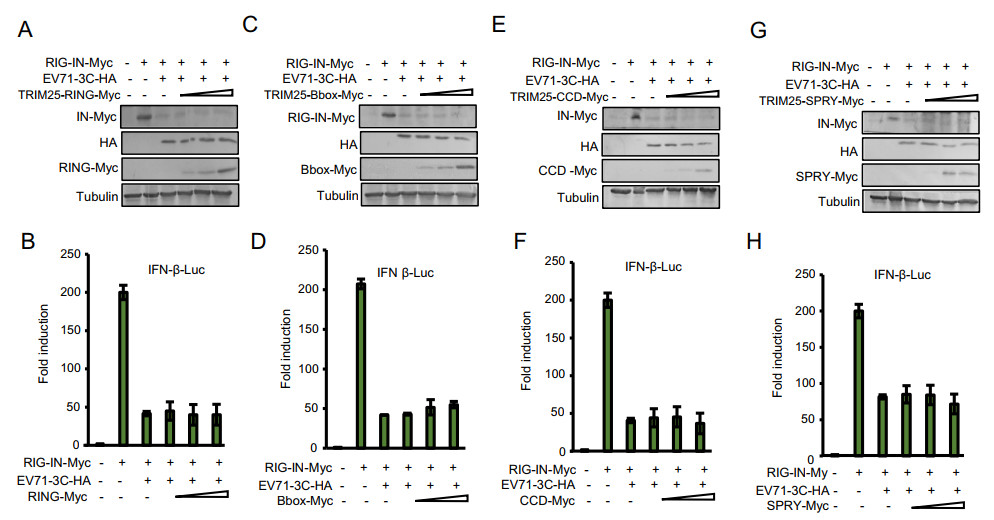
Figure 5. The RING, B-box, CCD or SPRY domain of TRIM25 alone can not restore the 3C-mediated inhibition of IFN-β response. A, C, E and G HEK293T cells were co-transfected with the IFN-β-Luc firefly luciferase reporter plasmid, Renilla luciferase control reporter plasmid pRL and 300 ng of RIG-IN, 300 ng of EV71 3C, increasing dose (50 ng, 150 ng, 450 ng) of TRIM25 mutant plasmids as indicated for 48 h, and then the cells lysates were subjected to immunoblotting with anti-Myc, anti-HA or anti-Tubulin antibodies. B, D, F and H Luciferase activities of the cell lysates were analyzed with the Dual-Luciferase Reporter Assay System and measured on a Monolight 2010 luminometer. Data represent the average of three independent experiments.
-
To explore whether the reduced RIG-I and TRIM25 expression caused by 3C is associated with its RNA binding and protease cleavage activity, three mutants H40G, E71A and C147G with a single amino acid substitution in EV71 3C were constructed. Increasing amount of EV71 3C WT, H40G, E71A and C147G were co-transfected with RIG-IN-Myc or TRIM25 expression vector into HEK293T cells (EV71 3C WT protein as a positive control, VR1012 as a negative control). After 48 h, the lysates from HEK293T cells were subjected to Western blot analysis. EV71 3C WT could induce the reduction of RIG-IN and TRIM25 in a dose-dependent manner (Fig. 6A and 6B), while the three mutants all lost the ability to reduce RIG-I or TRIM25 expression (Fig. 6C–6H).
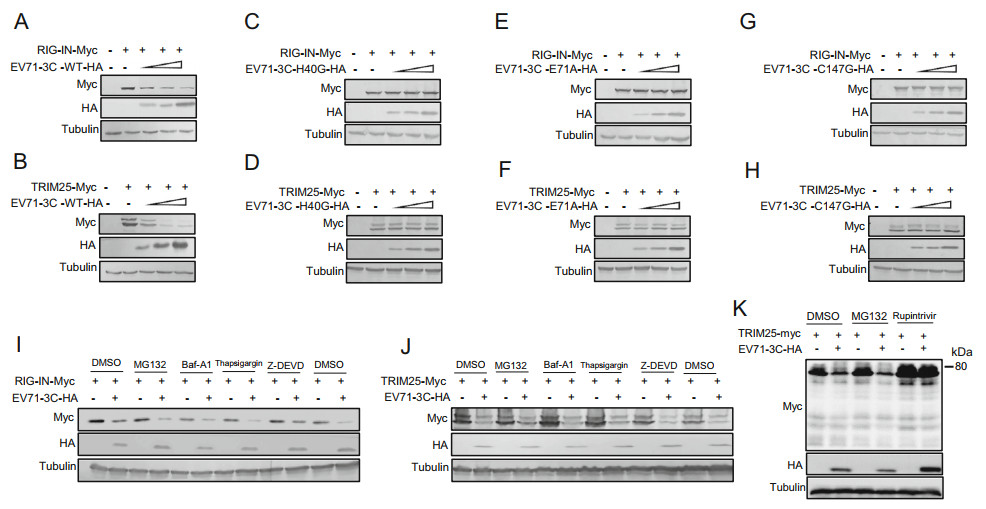
Figure 6. Cleavage activity of EV71 3C protein is required for reducing RIG-I and TRIM25 expression. A EV71-3C-H40G-HA (C), EV71-3C-E71A-HA (E) or EV71-3C-C147G-HA (6G) plasmids for 48 h, and then the cells lysates were subjected to immunoblotting with anti-Myc, anti-HA or anti-Tubulin antibodies. B, D, F and H Cell lysates from co-transfected HEK293T cells with indicated WT or mutant 3C and VR1012 or TRIM25 were subjected to immunoblotting with anti-Myc, anti-HA or anti-Tubulin antibodies. I, J and K HEK293T cells were co-transfected with RIG-IN-Myc and EV71-3C-HA plasmid. At 6 h post-transfection, Z-DEVD inhibitor was added for another 24 h according to the manufacturer's instructions. At 18 h post-transfection, MG132, Baf-A1, Thapsigargin inhibitor or Rupintrivir was added for another 24 h, and DMSO used as a negative control. The cell lysates were then subjected to immunoblotting with anti-Myc, anti-HA or anti-Tubulin antibodies.
To further investigate the mechanism of 3C inhibiting RIG-I and TRIM25 expression, various inhibitors blocking divergent pathways were used, MG132 for the proteasomal pathway, Baf-A1 for the lysosomal pathway, Thapsigargin for the autophage pathway, Z-DEVD for apoptosis and Rupintrivir for 3Cpro activity. HEK293T cells were co-transfected with EV71 3C and RIG-I or TRIM25 as indicated. According to the manufacturer's instructions, cells were treated with DMSO (as a negative control), MG132, Baf-A1, Thapsigargin inhibitor or Rupintrivir at 24 h post transfection, or treated with Z-DEVD inhibitor at 6 h post transfection. After another 8 h or 24 h, cells were harvested for Western blot analysis. As shown in Fig. 6I, 6J and 6K, none of the inhibitors but Rupintrivir could inhibit the 3C-mediated reduction of RIG-I and TRIM25, consistent with the conclusion that protease cleavage activity of EV71 3C is required for decreasing the expression of RIG-I and TRIM25.
-
A previous study reported that influenza A virus non-structural protein NS1 specifically inhibits TRIM25-mediated RIG-I ubiquitination and suppresses IFN production induced by RIG-I through competitively binding to the coiled-coil domain of TRIM25 (Gack et al. 2009). In order to examine whether EV71 3C protein blocks the interaction of RIG-I with TRIM25, we first detected whether 3C could bind to RIG-I or TRIM25. HEK293T cells were co-transfected with VR1012 or EV71 3C plus EV71 P1 or RIG-I and TRIM25 as indicated in Fig. 7A, EV71 P1 as a positive control. An anti-HA antibody conjugated to agarose beads was used to immunoprecipitate HA-tagged 3C from lysates of transfected HEK293T cells. The expression of 3C, EV71 P1, RIG-I and TRIM25 was determined by immunoblotting. However, RIG-I and TRIM25 failed to coimmunoprecipitate with 3C-HA, while EV71 P1 could (Fig. 7A). We also examined whether 3C could abrogate the interaction of RIG-I and TRIM25. As expected, RIG-I specifically coimmunoprecipitated with TRIM25-Myc but not with the sample containing the empty vector, VR1012, while addition of 3C had no effect on the interaction of RIG-I and TRIM25 (Fig. 7B).

Figure 7. EV71 3C has no effect on the interaction of RIG-I and TRIM25. A The interaction of EV71 with RIG-I and TRIM25. Cell lysates prepared from HEK293T cells expressing EV71 P1-Myc, TRIM25-Myc, RIG-I-Flag and EV71-3C-HA as indicated were coimmunoprecipitated with an anti-HA antibody conjugated to agarose and analyzed by immunoblotting with anti-Flag, anti-HA and anti-Myc antibodies. B EV71 3C has no effect on the interaction of RIG-I and TRIM25. Cell lysates prepared from HEK293T cells expressing indicated TRIM25-Myc protein and the RIG-I-Flag and EV71-3C-HA plasmids were coimmunoprecipitated with an anti-Myc antibody conjugated to agarose and analyzed by immunoblotting with anti-Flag and anti-Myc antibodies.
-
EV-D68, CVA6 and CVA16 all belong to the Picornavirus family. Although they have similar structures, they share only 70% to 80% homology with EV71. To affirm whether EV-D68, CVA6 and CVA16 3C proteins could suppress IFN-β expression via downregulating RIG-I and TRIM25, lysates from HEK293T cells co-transfected with VR1012 or divergent 3C proteins and TRIM25-Myc or RIG-I as indicated were subjected to Western blot analysis. Similar to EV71 3C protein, EV-D68 and CVA6 3C proteins downregulated RIG-I and TRIM25 expression in a dose-dependent manner (Fig. 8A and 8B). However, CVA16 3C protein could not suppress RIG-I and TRIM25 expression even at the maximum dose (Fig. 8C), suggesting that the CVA16 3C protein interferes with IFN-β production by another pathway. We further showed that overexpression of TRIM25 could rescue the RIG-I expression and restore IFN-β activation inhibited by EV-D68 and CVA6 3C proteins (Fig. 8D and 8E). In addition, we examined the effect of EV71, CVA6, EV-D68, and CVA16 infection on the expression of endogenous RIG-I and TRIM25 and found all viruses induced the production of RIG-I at the initial stage of infection. With the increasing infection time, the expression of RIG-I and TRIM25 was gradually reduced by EV71, EV-D68, and CVA6, but CVA16 did not significantly reduce the expression of RIG-I and TRIM25 (Fig. 8F–8I).
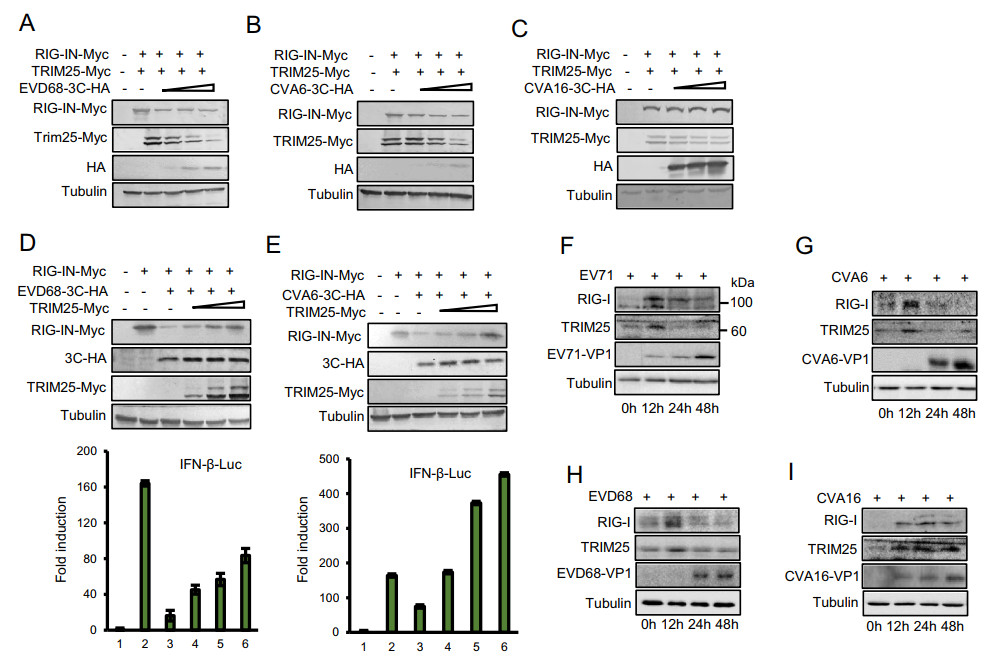
Figure 8. EVD68 and CVA6 but not CVA16 3C proteins suppress the IFN-β response via reducing RIG-I and TRIM25 expression. A, B and C HEK293T cells were co-transfected with the RIG-IN-Myc, TRIM25-Myc and EVD68-3C-HA (A), CVA6-3C-HA (B) or CVA16-3C-HA (C) plasmid. At 48 h post transfection, cell lysates were subjected to immunoblotting with anti-HA, anti-Myc or anti-Tubulin antibodies. D and E Overexpression of TRIM25 restored 3C-mediated reduced RIG-I expression and type Ⅰ IFN stimulation. HEK293T cells were co-transfected with the IFN-β-Luc firefly luciferase reporter plasmid, Renilla luciferase control reporter plasmid pRL and the indicated plasmids for 48 h. F–G The effect of EV71, CVA6, EVD68, CVA16 infection on RIG-I and TRIM25 expression. HEK293T cells were infected with indicated viruses for 48 h. The protein expression was then detected by immunoblotting using indicated antibodies. Luciferase activities of the cell lysates were analyzed with the Dual-Luciferase Reporter Assay System and measured on a Monolight 2010 luminometer. Data are representative of three independent experiments. Error bars indicate the SD of three different experiments.
3C Proteins of EV71 and CVB3 Inhibit IFN Signaling by Reducing Expression of RIG-I and TRIM25
Trim25 Rescues 3C-Mediated Inhibition on RIG-I Signaling Pathway
TRIM25 Stabilization of RIG-I Is Associated with Its Ubiquitination Function on RIG-I
Cleavage Activity of EV71 3C Is Required for Reducing RIG-I and TRIM25 Expression
EV71 3C Protein does not Disrupt the Interaction of TRIM25 and RIG-I
EV-D68 and CVA6 but not CVA16 3C Proteins Suppress IFN-β Response via Degrading RIG-I and TRIM25
-
As the first line of defense against virus infection, type Ⅰ IFN plays a critical role in initiating host antiviral responses. RIG-I is a key sensor for viral RNA in the cytosol and initiates a signaling cascade that leads to the establishment of IFN-mediated antiviral responses. Following its binding to viral RNA, RIG-I undergoes a conformational change through Lys63-type ubiquitination by the E3 ligase TRIM25 (Gack et al. 2007), allowing its N-terminal CARD domain to interact with the CARD domain of the downstream mitochondria-bound adapter MAVS (Sumpter et al. 2005; Gack et al. 2009; Xiao et al. 2011; Hu et al. 2017). Previous studies showed that EV71 3C inhibits type Ⅰ IFN responses by cleavage of IRF7 or TRIF (Lei et al. 2011, Lei et al. 2013). EV71 3C may also simply interact with the CARD region to preclude MAVS recruitment, and RIG-I fails to form a functional complex with MAVS and TBK1 without affecting levels of RIG-I or MDA5 in infected cells (Lei et al. 2010). However, Vincent et al. showed that RIG-I and MDA-5 are cleaved or degraded during picornavirus infection (Barral et al. 2007; Barral et al. 2009). Moreover, the ubiquitin ligase TRIM25 was demonstrated to be utilized by IAV NS1 protein to evade recognition by RIG-I (Gack et al. 2009). NS1 proteins encoded by human, avian, swine and mouse-adaptor influenza viruses all interact with TRIM25, thereby specifically inhibiting TRIM25-mediated RIG-I CARD ubiquitination that is required for antiviral activity (Rajsbaum et al. 2012). Therefore, the precise mechanism by which EV 3Cpro proteins inhibit RIG-I activation and whether TRIM25 plays a critical role in this process are worth to be further investigated. In this study, we demonstrate that the EV 3Cpro proteins could inhibit IFN-β production by specifically downregulating RIG-I and TRIM25 expression (Fig. 1 and 2). Overexpression of TRIM25 could rescue the RIG-I expression and restore IFN-β production reduced by 3Cpro proteins (Fig. 3). Thus, EV71 3Cpro protein-induced downregulation of TRIM25 and RIG-I may be a mechanism for antagonizing the innate response to viral infection.
Previous studies have shown that RIG-I CARD-dependent signaling is regulated by the dynamic balance between phosphorylation and TRIM25-induced K63-linked ubiquitination, while ubiquitination of RIG-I is critical for the ability of RIG-I to induce an antiviral IFN response (Gack et al. 2007). Acting as an E3 ubiquitin ligase, TRIM25 also is involved in MAVS ubiquitination and full NF-kB activation by ubiquitinating TRAF6 (Castanier et al. 2012; Lee et al. 2015). Subsequent studies showed that viral proteins interfere with TRIM25 function on RIG-I ubiquitination. For instance, SARS-CoV N protein was shown to suppress IFN-β production by targeting the SPRY domain of TRIM25 and disturbing the interaction between this SPRY domain and the N-terminal CARDs of RIG-I, resulting in the inhibition of RIG-I ubiquitination and downstream signaling pathway (Hu et al. 2017). Interestingly, our research indicated that EV 3C proteins could inhibit the RIG-I signaling pathway by downregulating RIG-I and TRIM25, which is crucial in the RIG-I-mediated activation of the type Ⅰ IFN pathway. Most human TRIM family proteins contain at least four domains (an N-terminal RING domain, one or two B-boxes, a central coiled-coil domain [CCD] and a SPRY domain), which are involved in broad biological processes, including diverse cellular functions as well as antiviral and antimicrobial activities (Nisole et al. 2005; Ozato et al. 2008; Munir 2010). Here, we demonstrated that two sites L69 and V72 in TRIM25 required for RIG-I ubiquitination could not restore the degradation of RIG-I induced by 3C protein (Fig. 4). Four single truncated mutants also could not restore the reduction of RIG-I expression (Fig. 5), suggesting that higher-order oligomerization of TRIM25 is structurally required for TRIM25 catalytic activation, which is consistent with the conclusion reported by Sanchez et al. (Sanchez et al. 2016).
Notably, the EV71 3C protein bearing proteolytic and RNA binding activities has been shown to be capable of inhibiting the RIG-I signaling pathway dependent on H40, KFRDI, and VGK motifs, whereas the protease and RNA binding activities are dispensiblee (Lei et al. 2010). However, our study showed that the single H40G, E71A or C147G substitution in the catalytic site significantly impaired the downregulation of RIG-I and TRIM25 compared to WT 3C (Fig. 6A–6H), suggesting that the cleavage activity of EV71 3C is closely associated with its inhibition of IFN production, although we didn't observe the cleavage band. At the same time, the various inhibitors including protease inhibitor and so on all could not rescue RIG-I and TRIM25 expression reduced by EV71 3C, further suggesting that the cleavage activity of 3C is important for RIG-I and TRIM25 reduction (Fig. 6J, 6K). Although EV71 3C can induce apoptosis by cleavage of some proteins (Li et al. 2002, 2017, 2019), the apoptosis inhibitor couldn't resue the expression of RIG-I and TRIM25 (Fig. 7I-7J), indicating that the apoptosis is not the reason that causes RIG-I and TRIM25 downregulation. Additional experiments showing that 3C could not disrupt the interaction of TRIM25 and RIG-I further support our conclusion (Fig. 7).
Except for the effect of EV71 3C on RIG-I and TRIM25, here we also showed that the 3Cpro proteins of EVD68 and CVA6 but not CVA16 decreased the expression of RIG-I and TRIM25 and inhibited IFN-β production. Overexpression of TRIM25 also restored the expression of RIG-I and IFN activation inhibited by 3Cpro of divergent EVs (Fig. 8A–8E). Our work provides in-depth insight into the mechanism of EV71-induced innate immune suppression. Thus, recovery of the RIG-I protein levels may be an efficient approach to activate the IFN signaling pathway through targeting TRIM25.
-
We thank C.Y. Dai for the critical reagents and P.T. Sarkis for editorial assistance. The study was supported by the National Natural Science Foundation of China (No. 81672004 and 81930062); the Science and Technology Department of Jilin Province (20190101003JH); and the Key Laboratory of Molecular Virology, Jilin Province (20102209).
-
BL and WZ participated in the study design and preparation of the manuscript; HX, JL, XY and ZL participated in the laboratory experimental work; YR participated in the analysis of the data. BL and ZY revised and finalized the manuscript. All authors read and approved the final manuscript.
-
The authors have declared that they have no conflict of interests.
-
This article does not contain any studies with human and animal subjects performed by any of the authors.

 DownLoad:
DownLoad: