












HTML
-
Human respiratory syncytial virus (RSV) is a major pathogen of acute lower respiratory tract infection (ALRTI) among young children (Nair et al. 2010; Shi et al. 2017), especially, the infants younger than five years of age. RSV infection can lead to hospitalization and death in children, especially in infants born prematurely, with congenital diseases, or immunocompromised (Welliver 2003). In 2015, models estimated that RSV caused about 33.1 million episodes of ALRTI worldwide, resulted in about 3.2 million hospital admissions, and 59, 600 in-hospital deaths in children younger than 5 years. Most of these deaths were in developing countries (Nair et al. 2010; Nolan et al. 2015; Shi et al. 2017). In China, RSV was also the majority viral pathogen in children aged < 2 years with ALRTI, which accounted for 17.0% (Feng et al. 2014).
As a member of the family Pneumoviridae, RSV is a non-segmented, negative-sense single-stranded RNA virus with an approximately 15.2 kb genome that encodes 11 structural and non-structural proteins (Canedo-Marroquin et al. 2017; Rima et al. 2017). The structural protein G is a glycoprotein, responsible for the viral attachment to the host cell. The genome of protein G contains two hypervariable regions (HVR-1 and HVR-2) in the ectodomain, while the C-terminal region exhibits the greatest genetic diversity and is widely used for the genotypes subdividing (Cane and Pringle 1995; Cane 1997). RSV is divided into RSV subgroup A (RSV-A) and B (RSV-B), which shows clear phylogenetic divergence (Mufson et al. 1985; Johnson et al. 1987). To date, at least 15 RSV-A genotypes [GA1-GA7 (Peret et al. 2000), SAA1 (Venter et al. 2001), CB-A(Baek et al. 2012), NA1-NA4 (Cui et al. 2013) and ON1-ON2 (Eshaghi et al. 2012; Gimferrer et al. 2019)] and 30 RSVB genotypes [GB1-GB4 (Peret et al. 2000), SAB1-SAB4 (Arnott et al. 2011), URU1-URU2 (Blanc et al. 2005), BA1-BA14 (Dapat et al. 2010; Baek et al. 2012; Etemadi et al. 2013; Bashir et al. 2017; Gimferrer et al. 2019), CB1 (Cui et al. 2013), GB5 (Ren et al. 2015), BA-C, BA-CCA, BA-CCB (Zheng et al. 2017), NZB1-NZB2 (Matheson et al. 2006), and CBB (Baek et al. 2012)] were reported.
Clarifying the molecular epidemiologic characteristic of the predominant RSV genotypes is important to obtain new insight into how RSV persistently causes frequent re-infections, epidemics and provide data for RSV prevention and vaccine development. The molecular epidemiology, circulation pattern, and evolution of different RSV genotypes of China before 2014 have been reported in previous studies (Song et al. 2017a; Song et al. 2017b). The results showed that ON1 replaced the NA1 as the predominant genotype of RSV-A during 2013-2015, while the BA9 was the main genotypes of RSV-B circulating between 2008 and 2014. However, the molecular epidemiology of RSV-A and -B genotypes after 2015 has not been studied extensively. The present multi-center prospective study was conducted to clarify the genetic diversity and circulating pattern of prevalent genotypes present in the mainland of China during 2015-2019.
-
From January 2015 to December 2019, respiratory samples were collected from patients hospitalized with community-acquired pneumonia (CAP) in a multi-center study that involved 14 hospitals of 11 provinces or municipalities (Beijing, Chongqing, Jilin, Hebei, Ningxia, Zhejiang, Guizhou, Liaoning, Shanghai, Heilongjiang and Guangdong). The eligible participants were younger than 18 years. Nasopharyngeal swabs were collected on admission and frozen at - 80 ℃. Total DNA/RNA from clinical samples were extracted by using QIAamp MinElute Virus Spin Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Multiplex real-time PCR assay was used to detect RSV-A, RSV-B, and other respiratory viruses (Multiplex Real-Time PCR Diagnostic Kit for Rapid Detection of 15 respiratory viruses; XABT, Beijing, China).
-
For RSV-positive samples, the HVR-2 sequence of RSV G gene was amplified by SuperScriptTM Ⅲ One-Step RT-PCR System (Invitrogen, Carlsbad CA, USA, cat: 12574018) using the primer pairs RSV-A-G-F/RSV-G-R (RSV-A-G-F: GAAGTGTTCAACTTTGTACC; RSV-G-R: CAACTCCATTGTTATTTGCC) and RSVB-G-F/RSV-G-R (RSVB-G-F: AAGATGATTACCATTTTGAAGT; RSV-G-R: CAACTCCATTGTTATTTGCC) (Peret et al. 1998; Zhang et al. 2007). These sequences were submitted to GenBank with the accession numbers from MW527490-MW528024 (Supplementary Table S1).
-
Multiple sequence alignments, pairwise distance and phylogenetic analyses were all conducted by using MEGA software (Version 6.06, Sudhir Kumar, Arizona State University, Tempe, AZ, USA). The genetic diversity and sequence identities were determined by BioEdit software (Version 7.2.5).
For the phylogenetic analysis, a total of 191 HVR-2 sequences from China (including 168 RSV-A and 14 RSV-B collected between January 2015 and December 2019, and 2 RSV-A and 7 RSV-B collected between 2008 and 2012), as well as 93 sequences worldwide with genotype information were retrieved from GenBank database (Supplementary Table S2). The phylogenetic tree was generated according to the neighbor-joining method, while the Kimura 2-parameter model was used. The bootstrap method with 1000 replicates was used to estimate the reliability of phylogenetic inference, while only the values over 70% were shown in each tree.
-
N-linked and O-linked amino acid glycosylation sites in the HVR-2 of G protein were predicted by the NetNGlyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/) and the NetOGlyc 4.0 server (http://www.cbs.dtu.dk/services/NetOGlyc/) respectively. The potential N-linked glycosylation site was defined as N-X-S/T (where X was not Pro); while the S/T residues were the potential O-linked sugar acceptors.
-
The data were analyzed using GraphPad Prism software (version 5.00, GraphPad, La Jolla, CA, USA). Differences in the co-detection rates of different age groups were evaluated by Chi-squared Test. A P-value of < 0.05 was considered indicative of a statistically significant difference between values.
Sample Collection and RSV Detection
Sequence Determination of RSV G Gene
Genetic Characterization and Phylogenetic Analysis
Glycosylation Analysis
Statistical Analysis
-
A total of 964 (17.44%, 964/5529) RSV positive samples were identified from 5529 enrolled patients between January 2015 and December 2019 from 14 hospitals located in Beijing, Shanghai, Chongqing, Jilin, Hebei, Ningxia, Zhejiang, Guizhou, Liaoning, Heilongjiang and Guangdong provinces. Over the total research period, RSV-A was predominant, except in 2016 (Fig. 1). The ages of these patients ranged from 1 month to 13 years with a median age of 3.5 years. The male-to-female ratio was 1.92:1 (634 boys and 330 girls).
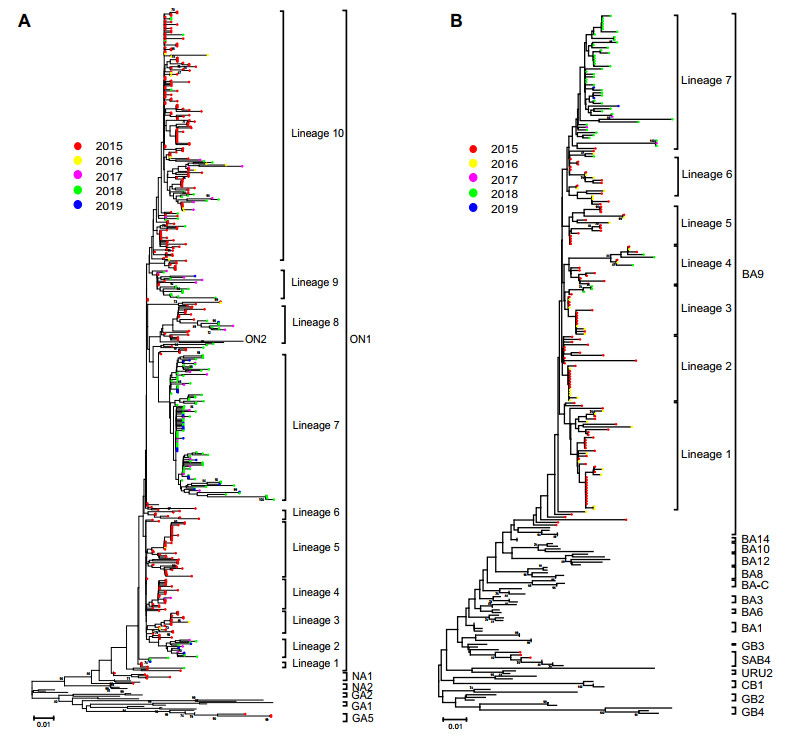
Figure 1. The phylogenetic dendrogram based on the Chinese RSV sequences from 2015 to 2019 and reference sequences with genotype information worldwide. A, B Detailed phylogenetic trees of the RSV-A and RSV-B taxa were analyzed, respectively. The neighbour-joining method was used to infer the topology by the MGEA 6.06 software. The percentage of bootstrap values (percentage of 1000 pseudoreplicate datasets) over 70% are shown at the nodes of the trees. Filled circles with different colors indicate the all the Chinese sequences from different years (red 2015, yellow 2016, purple 2017, green 2018, blue 2019). The scale bars represent substitutions per base pair per indicated horizontal distance.
Of these 964 RSV positive samples, 524 (54.36%) samples were identified as RSV-A, 183 (18.98%) samples were identified as RSV-B, 23 (2.39%) samples were identified as both RSV-A and B, and 234 (24.27%) samples were not tested due to insufficient specimen volume. Some other respiratory viruses were also detected in 311 (32.26%) RSV positive samples, while the co-detection rates were 37.60%, 29.51% and 34.78% in RSV-A, RSV-B and RSV-A & -B, respectively. The main respiratory viruses co-detected with RSV were Enterovirus/Rhinovirus (EV/HRV) (149, 15.46%), Bocavirus (HBoV) (72, 7.47%), Adenovirus (AdV) (49, 5.08%), Parainfluenza (PIV) (41, 4.25%), Human Metapneumovirus (hMPV) (40, 4.15%) and Influenza Virus (Flu) (40, 4.15%). The co-detection rates were 30.40% (176/579), 36.25% (95/262), 27.55% (27/98), and 52.00% (13/25) in the ages of ≤ 1 year, 1-3 years, 3-6 years, and > 6 years, respectively (P < 0.05).
-
A total of 535 RSV HVR-2 sequences of G gene was obtained in this study, including 353 RSV-A and 182 RSV-B. Combined with the 182 sequences (168 RSV-A and 14 RSV-B) retrieved from GenBank between 2015 and 2019, 717 HVR-2 sequences of G gene were performed molecular phylogenetic analysis (Table 1 and 2).
Province/Municipality Beijing Chongqing Jilin Hebei Ningxia Zhejiang Guizhou Liaoning Guangdong Fujian Gansu Total 2015 23 (23) 1 (1) 66 (0) 23 (23) 11 (11) 116 (116) 66 (28) 10 (0) 316 (202) 2016 2 (2) 5 (5) 9 (9) 16 (16) 2017 1 (1) 1 (1) 3 (3) 5 (5) 3 (3) 16 (16) 1 (0) 30 (29) 2018 16 (16) 1 (1) 31 (31) 29 (29) 1 (1) 53 (10) 131 (88) 2019 18 (18) 10 (0) 28 (18) Total 60 (60) 1 (1) 66 (0) 28 (28) 13 (13) 159 (159) 34 (34) 4 (4) 145 (54) 10 (0) 2 (0) 521 (353) Numbers shown are the sum of sequences collected in this study (shown in parentheses) and retrieved from GenBank database. Table 1. Distribution of RSV-A G gene HVR-2 sequences collected in the mainland of China from 2015 to 2019.
Province/Municipality Beijing Shanghai Chongqing Jilin Hebei Zhejiang Guizhou Liaoning Heilongjiang Guangdong Fujian Total 2015 7 (7) 3 (3) 14 (6) 43 (43) 27 (27) 2(2) 4 (4) 5 (0) 105 (92) 2016 5 (4) 1 (1) 3 (3) 20 (20) 21 (21) 50 (49) 2017 2 (2) 1 (1) 3 (3) 2018 6 (6) 2 (2) 13 (13) 1 (1) 22 (22) 2019 3 (3) 13 (13) 16 (16) Total 21 (20) 1 (1) 6 (6) 14 (6) 63 (63) 52 (52) 14 (14) 1 (1) 2 (2) 17 (17) 5 (0) 196 (182) Numbers shown are the sum of sequences collected in this study (shown in parentheses) and retrieved from GenBank database. Table 2. Distribution of RSV-B G gene HVR-2 sequences collected in the mainland of China from 2015 to 2019.
Phylogenetic analysis of 521 RSV-A sequences showed that 512 (98.27%) fell within genotype ON1 with the mean genetic distance of 0.029, 6 (1.15%) within genotype NA1 with the mean genetic distance of 0.052, and 3 (0.58%) within genotype GA5 with the mean genetic distance of 0.040. Almost all the RSV-B sequences clustered into BA9 genotype (193, 98.47%) with a mean genetic distance of 0.025; except three sequences were clustered into genotype SAB4 (mean genetic distance of 0.020). Only genotypes ON1 and BA9 were detected after December 2015 in this study.
For the genotype ON1, all the sequences after 2015 formed 10 lineages presenting temporal distribution. Most of the sequences from 2015-2016 were clustered together and formed lineage 1, 3-6, and 10; while most of the sequences from 2017-2019 formed lineage 2 and 7. Consistent with genotype ON1, the majority of BA9 sequences from 2017-2019 formed a new lineage 7, while these sequences from 2015-2016 separated into other 6 lineages (Fig. 1).
-
The deduced protein lengths of the RSV-A HVR-2 were 86 (genotypes NA1), 87 (genotypes GA5) and 110 (genotype ON1) amino acids, respectively. All the RSV-A sequences from China between 2015 and 2019 were analyzed to determine the amino acid variations (Fig. 2). Seventy-three amino acid changes were detected in the 512 sequences of HVR-2 of genotype ON1 compared with the reference (ON67-1210, GenBank accession number: JN257693). For the frequency of the amino acid changes, 35 sites were more than 1%, however, other 38 sites with frequency less than 1% were not shown. Only 13 sites more than 5% were identified [V225A (8.20%), T245A (5.27%), T249I (30.47%), H258Q (23.83%), E262K (38.28%), H266L (23.83%), E271K (6.25%), L274P (17.97%), E286K (6.45%), Y297H (7.42%), L298P (18.36%), Y304H (8.59%), L310P (5.86%)], which contained 6 sites more than 10% in them (Fig. 2). All the 196 sequences with amino acid change E262K clustered into lineage 10, including all the 156 sequences with amino acid changes T249I. For the 196 sequences, 161, 12, 6 and 17 of them were from 2015, 2016, 2017 and 2018 respectively. Almost all the sequences with amino acid changes H258Q and H266L were from Beijing, Zhejiang, Ningxia, Guangdong and Jilin municipality/provinces between 2017 and 2019, except 5 from Zhejiang and Fujian provinces in 2015. The two substitutions were co-occurrences, while all sequences with these two substitutions clustered into lineage 7 (Fig. 1).

Figure 2. Summary of amino acid changes and frequencies in HVR-2 of the G gene of genotype GA5 (n = 3), NA1 (n = 6), ON1 (n = 512), SAB4 (n = 3), and BA9 (n = 193) comparing with their reference sequences. The earliest sequence of each genotype was used as the reference (GA5: Mon/1/01, AF516135; NA1: NG-016-04, AB470478; ON1: ON67-1210, JN257693; SAB4: Cam2009-0351, JN119976; BA9: BA/100/04, DQ227395). Only sites with a substitution frequency of more than 2% are indicated. For the RSV-A ON1 and RSV-B BA9 genotypes, these substitution frequency more than 5% or 10% were marked with orange and red label, respectively.
For the genotype NA1, there were 10 specific amino acid changes compared with the reference sequence (NG-016-04, GenBank accession number: AB470478) from Japan in 2004. Specific amino acid substitutions were P222S (66.67%), D237N (100%), N251Y (50.00%), N260S (100%), T269S (33.33%), N273Y (83.33%) and L274P (33.33%). Compared with the reference sequence (Mon/1/01, GenBank accession number: AF516135) from Uruguay in 2001, there were 7 amino acid changes in the genotype GA5, including E224D (66.67%), T227I (66.67%), E232D (66.67%), T243I (100%), S267L (33.33%) (Fig. 2).
The amino acid variations analyses were also performed on the HVR-2 regions of RSV-B protein G between 2015 and 2019 (Fig. 2). There were 5 and 44 amino acid substitutions in SAB4 and BA9, compared with their respective reference sequences (Cam2009-0351, GenBank accession number: JN119976 and BA/100/04, GenBank accession number: DQ227395). For the amino acid changes frequency of the genotype BA9, 14 sites were more than 5% and 9 sites were more than 10% (Fig. 2). Compared with the reference (DQ227395), the deduced 20 amino acid duplicated regions of all genotype BA9 sequences have 10 amino acid substitutions, containing R262G (2.07%), P264T/A (98.45%, 1.55%), L267S/P (98.45%, 1.55%), I270T (3.63%), A271V (16.06%), T275A (2.07%), T276A (5.18%), S269F (1.04%), L272P (1.55%) and D273N (1.55%) (Fig. 2). All the sequences with amino acid changes T290I and T312I were from 2018 and 2019, which clustered into lineage 7 (Fig. 1). The amino acid change P237H (8.81%) was only observed in 17 sequences from Hebei Province between 2015 and 2016, which belonged to the lineage 1 (Fig. 1). For the genotype SAB4, specific amino acid substitutions were found at P216Q (33.33%), T229A (66.67%), E258K (66.67%), I280T (100%) and F287S (33.33%).
-
N-X-S/T, where X was not Pro, was identified as the N-glycosylation site. Three predicted N-glycosylation sites (237, 250 and 294) were relatively conserved among the genotype GA5 between 2015 and 2019. For the genotype NA1, three predicted potential N-glycosylation sites were located at 237, 251 and 294, while N251Y amino acid substitution was identified in 50% of the sequences, respectively. In contrast, only one N-glycosylation site 237 was identified among the genotype ON1 sequences. For the RSV-B, two N-glycosylation sites were identified and conserved among all the sequences of genotypes SAB4 and BA9 in this study. The sites were 276 and 290 for the genotype SAB4, while for the genotype BA9 sequences the sites were 296 and 310.
The protein G also contains numbers of serine and threonine residues which are potential O-glycosylation sites (Wertz et al. 1985). Analysis of the HVR-2 predicted 2-10 potential O-glycosylation sites among genotype ON1 sequences, and 12-24 potential O-glycosylation sites among genotype BA9 sequences between 2015 and 2019 (G-scores: 0.5-0.9) (Supplementary Table S3 and S4). The amino acid positions most likely to have O-glycosylation are T219, T220, T227, T228, T231, S270, S275, S277, T281, S283 for genotype ON1 (refer to GYYF-57), and T218, T222, T227, T228, T232, T236, T240, T244, S245, T246, S249, T250, T255, S257, T260, S265, T280, T281, S285, T294, S297, T298, T300, S304 for genotype BA9 (refer to ZJ-293).
Sample Information
Phylogenetic Analysis of RSV G Gene HVR-2 Sequences
Variation of the Deduced Amino Acid Sequence
Glycosylation Sites of the Deduced Amino Acid Sequence
-
RSV is an important pathogen of global distribution. It is the most frequently identified agent of ALRTI in children, especially in hospitalized patients younger than 5 years. For the paucity of information on molecular epidemiology, the present multi-center prospective study was conducted to investigate the genetic diversity and circulating pattern of prevalent RSV genotypes present in the mainland of China during 2015-2019. A total of 5529 samples from CAP patients were collected, and 964 (17.44%) of the enrolled patients were identified to be positive for RSV.
There are conflicting reports regarding the associations of different groups and genotypes with the severity of RSV infection (Yoshihara et al. 2016; Laham et al. 2017; Otieno et al. 2017; Vandini et al. 2017). Due to lacking enough clinical feature records, the data in this study were also not sufficiently conclusive for considering the association between genotypes and the severity of disease. The co-infection of RSV with other respiratory viruses is common in many hospitalized children with ALRTI. The clinical implications of multiple viral infections remain controversial. Some studies showed that multiple respiratory virus co-infection might increase the clinical severity of RSV disease, whereas other studies suggested no influence on clinical manifestation (Richard et al. 2008; Franz et al. 2010; Papenburg et al. 2012; Harada et al. 2013; Mazur et al. 2017). It warrants further study about viral co-infection. In this study, co-infection with RSV and other respiratory viruses was detected in 311 hospitalized children, which accounted for 32.26% of all the RSV associated ALRTI. The co-detection rate of RSV-A with other respiratory viruses was slightly higher than the rate of RSVB (P < 0.05). The main co-infected viruses were EV/HRV, HBoV, AdV, PIV, hMPV and Flu. The co-detection rate was 52.00% in the patients older than 6 years, which was significantly higher than other age groups.
The genotypes ON1 and BA contained 72 and 60 bp duplication in the HVR-2 of the G gene (Trento et al. 2006; Eshaghi et al. 2012). The ON1 had become the predominant genotype and spread all over the world after it was first detected in Canada in 2010. In China, the genotype ON1 became more and more prevalent since detected in Shanghai in February 2011, and replaced the genotype NA1 to become the predominant RSV-A genotype during 2013-2015 (Song et al. 2017b). The first BA variant was isolated in Madrid in 1998. Then, the genotype BA had spread globally and evolved into 14 genotypes (Trento et al. 2010; Gimferrer et al. 2016; Abrego et al. 2017). After first detected in 2006, the genotype BA9 became the predominant genotype of RSVB in China during 2008-2014 (Song et al. 2017a). In this study, genotypes ON1 (307), NA1 (6), GA5 (3) of RSV-A and genotypes BA9 (102), SAB4 (3) of RSV-B were found in 2015, while the ON1 and BA9 were the predominant genotypes accounting for 97.15% and 97.14%, respectively. After December 2015, genotypes ON1 and BA9 had completely replaced other RSV genotypes. The lengthening modification in G glycoprotein probably reflects an evolutionary advantage, which may be the reason for the spreading and replacing of genotypes ON1 and BA9.
The protein G, as an attachment protein, plays a critical role in virus entry into the host cells. It can affect human immune responses and facilitate viral evasion, which is also a significant candidate for RSV vaccine and antibody development (Chirkova et al. 2013). The variations at the protein G are also the marker to trace the origin of infection. Therefore, it is of great importance to identify the amino acid variations of the circulating genotypes.
Compared with the reference sequence (ON67-1210), six sites were identified with the frequency of the amino acid changes more than 10% in the sequences of HVR-2 of genotype ON1 between 2015 and 2019. The amino acid substitutions T249I and E262K have been reported in the previous study (Song et al. 2017b), while the H258Q (23.83%), H266L (23.83%), L274P (17.97%), L298P (18.36%) are reported for the first time. Interestingly, all the sequences with amino acid changes H258Q and H266L were co-occurrences, and clustered into the same lineage. The T290I and T312I were new amino acid changes in genotype BA9 between 2018 and 2019, while all the sequences with these two changes clustered into a new lineage. The amino acid change P237H (8.81%) had certain geographical limitations, which were observed only in 17 sequences from Hebei Province between 2015 and 2016.
The virus evasion from host immune response recognition may be mediated by the presence of N- and O-linked glycans on the G protein. The antigenicity of RSV protein G can be severely affected by the pattern and frequency of N- and O-linked carbohydrate side chains (Palomo et al. 1991; Khan et al. 2015; Haider et al. 2018). For the predominant genotype ON1, only one conserved N-glycosylation site was identified in position 237. All the genotype BA9 in this study have two conserved N-glycosylation sites at position 296 and 310 of the 60-nucleotide duplication region. These results showed that N-linked glycosylation sites on the protein G did not affect the antigenic alterations of RSV.
In conclusion, this is a multi-center molecular epidemiologic study of the RSV in China between 2015 and 2019. The result suggested that the ON1 and BA9 were the predominant genotypes for RSV-A and RSV-B respectively. Meanwhile, only ON1 and BA9 genotypes have been detected after December 2015 in this study. For the genotypes ON1 and BA9, the G gene exhibited relatively high diversity and could be separated into 10 and 7 lineages, respectively. The temporal and geographical factors may play a role in RSV genetic evolution. For the rapid evolution and widespread transmission of this virus, continued surveillance needs to be conducted to illustrate the molecular characterization of the emerging genotypes of RSV.
-
This work was supported by the National Science and Technology Major Projects (Grant Number 2017ZX10104001-005-010, 2017ZX10103004-004) and the CAMS Innovation Fund for Medical Sciences (CIFMS) (Grant Number 2019-I2M-5-026). These sponsors had no role in the study design, data analysis, manuscript preparation, or decision to publish.
-
XC and ZX conceived and designed the experiments. XC, YZ, WW and MZ performed the experiments. XC, CL, SA, GL, RJ, BX, YZ, AC and LL contributed specimens and analyzed the data. XC and ZX contributed to the writing of the manuscript. All authors reviewed the manuscript.
-
The authors declare that they have no conflict of interest.
-
This study was approved by the Medical Ethics Committee of the Beijing Children's Hospital, affiliated to Capital Medical University and informed consent was obtained. All procedures were conducted according to the guidelines (ethics number 2014-99 and 2017-k-15).

 DownLoad:
DownLoad: