











-
Infectious Hypodermal and Hematopoietic Necrosis Virus (IHHNV) and Taura Syndrome Virus (TSV) are two major causes of shrimp disease which were listed as the significant diseases by OIE (World organization for animal health). IHHNV was first found in Penaeus stylirostris from Hawaii in 1981 (10, 11) and caused mortalities of up to 90%. Since then, the virus has been detected in a number of other penaeid species around the world, including the Americas, Oceania, and Asia (5). IHHNV is a small, icosahedral, nonenveloped virus containing a single-strand linear DNA of approximately 4.1 kb. Based on size, morphology and biochemical structure (2, 13, 14, 15) IHHNV is considered to be a member of the family Parvoviridae. The IHHNV genome contains 3 open reading frames: ORF1 and ORF2 encoding putative non-structure proteins and ORF3 encoding a putative structure protein. TSV was first discovered in Ecuador in 1992 (8) and was the most important shrimp virus in western-hemisphere. The full-length genome of TSV has been sequenced and consists of a linear, positive, single-strand RNA of 10 205 nt which includes two open reading frames: ORF1 encoding putative non-structure proteins and ORF2 encoding capsid proteins VP1, VP2 and VP3 (1). According to its morphology and genome, this virus was classified into a new family Dicistroviridae.
The IHHNV and TSV were detected in China in 2002 and 2000 respectively and were believed to be introduced to China through importation of shrimp brood stock (3, 6, 12, 18). However, there is little information about the prevalence and genetic variation of these two viruses in China. In this study, we investigated the prevalence of these two viruses in several regions of China and present our findings regarding their genetic variation.
HTML
-
The samples of Penaeus vannamei were collected from seven different areas of China and were stored at -80ºC. Detailed information is given in Table 1.

Table 1. Samples information
-
For IHHNV detection, shrimp gills were homogenized in TN buffer (0.02 mol/L Tris-HCl, 0.4 mol/L NaCl, pH7.4) and then centrifuged at 6 000×g for 8 min. After discarding the pellet, the supernatant was boiled for 5min and then centrifuged at 10 000×g for 10 min, the final supernatant was used as the template for PCR amplification.
For TSV detection, the total RNA was extracted from the gills of samples with TRIzol reagent (Invitrogen, Carlsbad, USA) according to the manufacturer, s protocol. The final RNA was resuspended in 40 to 50 µL DEPC water and stored at -80 ºC.
-
The primers for IHHNV and TSV detection were designed using Primer Express software (Version 2.0) based on the IHHNV (GenBank AF218266) and TSV (AF277675) sequences, respectively. All primers used for IHHNV and TSV detection and genome fragment amplification are shown in Table 2.

Table 2. Primers used for IHHNV and TSV detection and amplification
Reverse transcription for TSV RNA was performed in a 25µL reaction which contained 2 μL total RNA, 100 pmol reverse primer (TDP02) and 10.5 μL of diethylpyrocarbonate-treated water. The reaction was first denatured at 70 ºC for 10 min, then immediately quenched on ice, and subsequently added to the RT mixture consisting of 1.25µL dNTPs (10 mmol/L), 5µL 5×M-MLV buffer, 25U RNasin (BioStar, CANADA) and 200U M-MLV reverse transcriptase (Promega, MADISON, WI, USA). The reverse transcription reaction was conducted at 42 ºC for 60 min, followed by heating to 70℃ for 5 min and holding at 4℃.
The PCR amplification for IHHNV and TSV was carried out in a 25µL reaction containing 1µL extracted DNA (for IHHNV) or 1µL cDNA (for TSV), 10 pmol of each primer, 0.5µL dNTPs (10 mmol/L), 2.5µL 10×Taq buffer, 0.5U of Taq polymerase (BioStar, CANADA). Amplification was performed with the following profile: 95℃ 5min, 30 cycles of 94℃ 30s, 53℃ or 55℃ 30s, 72℃ 35s, with a final extension for 72ºC 10 min, then incubate at 4℃ 5 min. The PCR product was analyzed by electrophoresis (100V, 30 min) in 1% agarose gels stained with ethidium bromide.
-
A 3 065 bp fragment (corresponding to nucleotides 792 to 3 856 of IHHNV isolates from Hawaii, GenBank AF218266) which contained the whole of ORF1 and ORF3 was amplified and sequenced from positive IHHNV samples. The whole of ORF2 (coordinate nucleotide 6 673 to 10 165 of TSV isolates from Hawaii, GenBank AF277675) was amplified and sequenced from TSV positive samples. The IHHNV and TSV sequences were compared with related sequences in GenBank (Table 3). Multiple alignments were performed with the Clustal X (version 1.83) software. Phylogenetic tree was calculated with the Mega (version 3.1) package under the NJ method using 1000 bootstrap replicates. Bootstrap values greater than 50% were shown.

Table 3. Information for IHHNV and TSV samples used in this study
Samples
DNA and RNA extraction
Samples detection
Sequence analysis
-
A total of 214 samples from seven different areas of China were detected using PCR or RT-PCR for both IHHNV and TSV. The results are summarized in Fig.1-3 and Table 4.
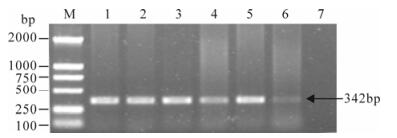
Figure 1. PCR detection for IHHNV using primers 77012F and 77353R (only positive samples are shown). M, DNA marker; 1-6, Shrimp samples; 7, Healthy crayfish used as negative control
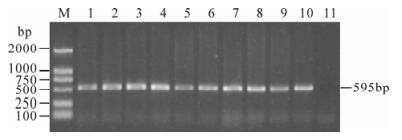
Figure 2. PCR detection for IHHNV using primers IDP03 and IDP04 (only positive samples were shown). M, DNA marker; 1-10, Shrimp samples; 11, Healthy crayfish used as negative control
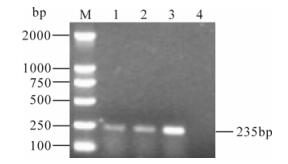
Figure 3. RT-PCR detection for TSV using primers TDP01 and TDP02 (only positive samples are shown). M, DNA marker; 1-3, Shrimp samples; 4, Healthy crayfish used as negative control

Table 4. Results of detection for IHHNV and TSV
The results showed that the prevalence of IHHNV in collected samples was much higher (65.42%) compared to TSV (3.27%) and some of the shrimp (2.34%) were co-infected with both viruses, indicating that these two viruses were present in the same host. Also, we observed that the percentage of IHHNV positive samples were different among different areas. The samples from Haikou, Guangdong and Zhanjiang have a much higher percentage of IHHNV (80%, 100% and 100%, respectively), then from Wenling, Zhongshan and Zhuhai (50%, 30% and 38%, respectively), with the fewest occurrences in samples from Ezhou (20%). Thus, it seems that there is a much higher prevalence of IHHNV in coastal areas (especially in Guangzhou and Zhanjiang) than inland areas (Ezhou).
-
A 3 065 bp fragment was amplified from IHHNV positive samples from 7 different locations. When compared with the Hawaii strain, a low variation was observed in both ORF1 and ORF3 and no deletions or insertions were detected. In total, there were 21 nucleotide substitutions were found within 2001 nt (1.00%) in ORF1 which resulted in 12 amino acid changes (1.80%), and 19 nucleotide substitutions within 990 nt (1.92%) in ORF3 which resulted in 10 (3.04%) amino acid changes (Table 5 and Table 6). The variation percentage is little higher in the capsid protein than in the non-structure protein for both the nucleotide and the amino acid sequences as previously reported (17).

Table 5. Nucleotide changes in non-structure protein ORF1 and structure protein ORF3

Table 6. Amino acid changes in non-structure protein ORF1 and structure protein ORF3. The IHHNV genome from Hawaii (GenBank accession No. AF218266) was used as a reference.
A 3493bp fragment which encodes the structural protein ORF2 was amplified from TSV positive samples from three different areas of China. The sequence identities of these isolates was between 97.8% to 98.3% when compared with the TSV Hawaii strain (GenBank accession No. AF277675). The distribution of the nucleotide and amino acid variation is shown in Table 7 and Table 8.

Table 7. Nucleotide and amino acid variations of TSV isolated from China compared with TSV isolated from Hawaii (GenBank accession No. AF277675).

Table 8. Mutation sites between Hawaiian isolate and Asian isolates. (numbers indicate the amino acid positions in TSV ORF2)
The result demonstrated that the variations for TSV are rather low and concentrated in VP1 region for both nucleotide and amino acid, suggesting that this region is prone to mutant.
-
The obtained sequence of IHHNV (ORF1 and 3) and TSV (ORF2) for the Chinese isolates were used for phylogenetic analysis. (Fig. 4-6).
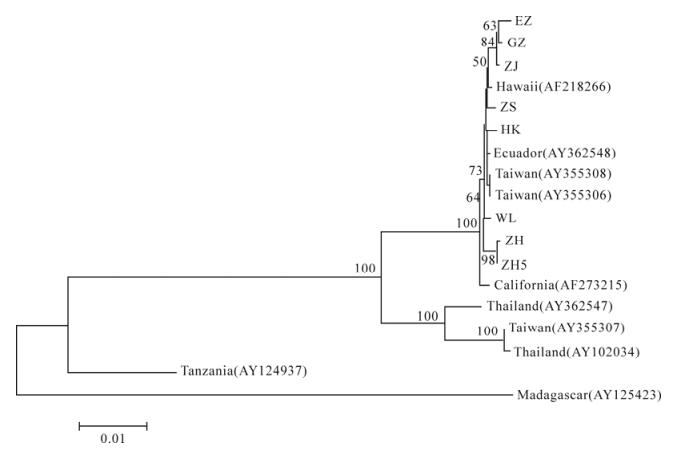
Figure 4. Phylogenetic tree based on IHHNV ORF1 nucleotide sequences. The phylogenetic trees were constructed using the Neighbor-Joining algorithm in the MEGA3.1 software with a bootstrap of 1 000 replicates. The number on the branches indicates bootstrap values >50. ZH5, isolate collected from Zhuhai and co-infected with IHHNV and TSV. EZ, from Ezhou; GZ, from Guangzhou; ZH, from Zhanjiang; ZS, from Zhongshan; HK, from Haikou; WL, from Wen lin; ZH, from Zhuhai.

Figure 5. Phylogenetic tree based on IHHNV ORF3 nucleotide sequences. The phylogenetic trees were constructed using the Neighbor-Joining algorithm in the MEGA3.1 software with a bootstrap of 1000 replicates. The number on the branches indicates bootstrap values >50. ZH5, isolate collected from Zhuhai and co-infected with IHHNV and TSV. EZ, from Ezhou; GZ, from Guangzhou; ZH, from Zhanjiang; ZS, from Zhongshan; HK, from Haikou; WL, from Wen lin; ZH, from Zhuhai.
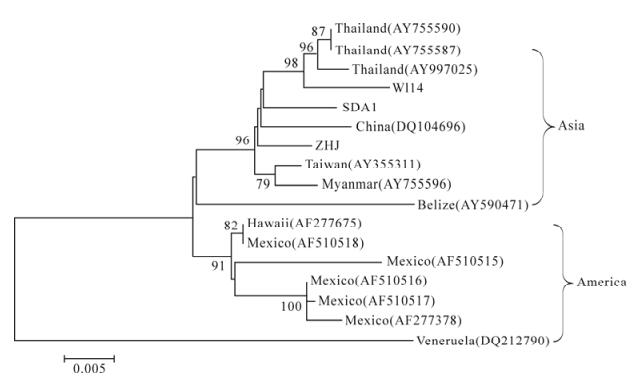
Figure 6. Phylogenetic tree based on TSV ORF2 nucleotide sequences. The phylogenetic trees were constructed using the Neighbor-Joining algorithm in the MEGA3.1 software with a bootstrap of 1000 replicates. The number on the branches indicates the bootstrap values >50. SDA1, from Shunde; WL14, from Wenlin; ZHJ, from Zhanjiang.
Results for IHHNV showed that the trees for both ORF1 and ORF3 were similar. All seven Chinese isolates were clustered with isolates collected from Hawaii (AF218266), Taiwan (AY355308 and AY-355306), Ecuador (AY362548) and California (AF-273215). Two Thailand isolates (AY362547 and AY102034) were grouped with a Taiwan isolate (AY355307). Two Africa IHHNV-related sequences (AY124937 and AY125423) were indicated to be distantly related to other IHHNV sequences. This result indicated that there are no apparent associations between IHHNV sequence variations and geographic origins.
Phylogenetic analysis of TSV revealed that the TSV isolates can be divided into two groups (Asia group and America group). Three Chinese isolates were clustered in the Asia group and showed a closer relationship with another Chinese isolate (DQ104696) collected from GenBank. This result demonstrated that the sequence variation of this virus was related to the geographic origins.
Moreover, we observed some mutations between the Hawaii isolate and all Asia isolates in the analysed sequence, suggesting that these variation sites may act as the genetic markers of Asian TSV isolates (Table 8), as suggested by Chang et al (4).
Samples detection
Sequence variations
Phylogenetic analysis
-
IHHNV and TSV are considered to be two important pathogens in America. However, with the frequent trade of brood stock, these two viruses were transmitted to Asia. Our results confirmed that there was a high prevalence of IHHNV in the shrimp farm where the samples were collected. All IHHNV positive samples showed no apparent symptoms, indicating that this infection was latent. This may be attributed to the development of a balanced hostpathogen relationship. Although IHHNV infection did not cause severe mortalities in cultured shrimp in recent years, the risk of an IHHNV outbreak in the future can not be excluded as long as the climate or environment change favor the virus replication in shrimps. Thus it is important to monitor and prevent this virus outbreak in future cultured shrimp.
Compared with IHHNV, the collected samples had a low prevalence for TSV which was only detected in three locations (Zhuhai, Wenling and Zhanjiang). All TSV positive samples showed no apparent symptoms. It was also interesting that we found several samples co-infected with IHHNV and TSV. Whether these two viruses were co-infected in the same cells needs to be further investigated through in-situ hybridization. In addition, we also detected WSSV by PCR in our collected samples and all were negative (data not shown).
Phylogenetic analysis indicated that the sequence variations for IHHNV and TSV are very low, with the exception of IHHNV-related sequences from Africa and Australia. The IHHNV-related sequences were demonstrated to be one part of the shrimp genome sequence and its origin and the relationship with IHHNV remains to be elucidated. These results implied that the IHHNV and TSV were well established in shrimp and have constantly evolved through the years. The VP1 sequence of TSV has higher nucleotide variations than other regions. There are nine different variation sites between the Asia and America TSV VP1 sequence, suggesting that these sites could be used as genetic markers of TSV from different origins.

 DownLoad:
DownLoad: