











-
Baculoviruses comprise a large family of invertebrate pathogenic viruses, infecting primarily insect species of the order Lepidoptera. They are considered to be safe biological insecticides with great potential in pest control. The family consists of two genera, nucleopolyhedroviruses (NPVs) and granuloiruses (GVs) (26). NPVs can be further characterized, depending on the number of nucleocapsids surrounded by a common membrane, into multiple (MNPV) and single (SNPV) nucleocapsid NPVs. On the basis of single-gene and genomic phylogenies the lepidopteran-specific NPVs have been divided into two groups, group Ⅰ NPVs and group Ⅱ NPVs (4, 6, 30). This subdivision also correlates with the presence of unique envelope fusion proteins GP64 (Group Ⅰ) and F (Group Ⅱ) encoded by viruses from each group (10, 21).
Fourty-one baculovirus genomes have been fully sequenced and characterized and new whole genome sequences are being published regularly (GenBank June 2007). Genome sequence analyses revealed that baculovirus phylogeny follows the classification of the host insect (7) and that morphological traits of e.g. polyhedra can be misleading (13). Recently, a new classification and nomenclature for the family Baculoviridae, including four genera (Alpha-, Beta-, Gamma-and Deltabaculovirus) have been proposed (13). At present, more than 700 baculoviruses have been reported (20) and many are considered or actually used as biocontrol agents of pest insects. Most notable are the baculoviruses of Anticarsia gemmatalis and Helicoverpa armigera for the control of the soybean looper and cotton bollworm at a large scale in Brazil and China, respectively (20, 31). Despite of their large number only a small subset of baculoviruses has been studied in detail.
Most phylogenetic analyses so far have been based on single-gene sequences (4, 30), which often led to conflicting results when different genes were examined. Combined phylogenies based on more than one gene have shown to alleviate this problem and to make the phylogenetic analyses more robust (6, 14, 16). Recently a criterion for distinguishing virus species has been proposed. The evolutionary distance between a pair of sequences usually is measured by the number of nucleotide (or amino acid) substitutions occurring between them. One of the models used to estimate the evolutionary distance between sequences is the Kimura 2-parameter, which corrects for multiple hits, taking into account transitional and transversional substitution rates and assuming that the four nucleotide frequencies are the same and that rates of substitution do not vary among sites. The proposed criterion suggests that when the Kimura 2-parameter distance between single or concatenated genes is larger than 0.05, two viruses may be considered as different virus species (14).
Two different nucleopolyhedroviruses naturally occur in populations of the Douglas-fir tussock moth, Orgyia pseudotsugata (Lepidoptera: Lymantriidae) (9), an important pest of interior Douglas-fir (Pseudotsuga menzesii var. glauca (Beissn.) and several species of true firs (Abies spp.) in North America (3). OpMNPV, the bioactive ingredient of the bioinsecticide TMBiocontrol-1 (registered both in the United States and Canada) is used to control the tussock moth in North America (22). Its genome has been completely sequenced, and the function of many genes has been determined (1, and references therein). OpSNPV was also originally isolated from O. pseudotsugata baculovirus isolates and is less virulent than OpMNPV. Both viruses are often found intermixed in the same insect population (8). The current hypothesis is that OpMNPV was introduced in the 1920s from Europe by Leucoma salicis larvae (11) and partially displaced OpSNPV. In Europe L. salicis NPV was probably an ancestral variant of OpMNPV, but names after its European host, Leucoma (Stilpnotia) salicis (LesaNPV) (11, 32). Both OpSNPV and OpMNPV show distinct restriction enzymes profiles (23). Phylogenetic analysis of the polyhedrin genes showed that OpMNPV belongs to group Ⅰ NPVs, while OpSNPV was ambiguously assigned as a group Ⅰ (28) or group Ⅱ NPV (4, 30). Relative to OpMNPV much less analysis has been done on OpSNPV, as to date only its polyhedrin gene has been studied (17, 18, 24).
The polyhedrin gene encodes the matrix protein of the NPV occlusion body and is one of the most conserved baculovirus genes (27). Initially, baculoirus phylogenies were created based on polyhedrin gene sequences due to the high number of such sequences available. The subdivision of lepidopteran baculoiruses in group Ⅰ and Ⅱ was made based on this protein (30). Although this grouping has been confirmed recently by gene content, gene order and whole genome phylogenies (5, 7), the polyhedrin gene phylogenetic analyses often gave conflicting results when compared to the phylogenies of multiple genes or genomes. Autographa californica MNPV, is the type species of group Ⅰ NPVs, while its polyhedrin gene falls out of group Ⅰ and belongs to group Ⅱ NPVs (12). This suggests that phylogenies based on single genes can give misleading results and that multiple gene sequence data are preferred for baculovirus characterition. Detailed analysis using models that can detect recombination events, revealed that the AcMNPV polyhedrin gene is in fact a mosaic of group Ⅰ and group Ⅱ NPV-specific polyhedrin sequences (12).
OpSNPV was included in only one of the gene phylogenies (7), since further gene sequence information of OpSNPV was lacking. In the present study we sequenced four conserved baculovirus core genes from OpSNPV and constructed a phylogenetic tree based on these four core gene sequences and resolved the phylogenetic position of OpSNPV.
HTML
-
In this study we amplified four conserved baculovirus genes from OpSNPV in PCR reactions using degenerate primer sets for polyhedrin (polh), late expression factor 8 (lef-8), per os infectivity factor 2 (pif-2) and DNA polymerase (dpol), respectively. The degenerate primer set for the polh gene was previously described by Moraes and Maruniak (19), and for the lef-8 and pif-2 genes by Herniou et al. (6, 8) (Table 1). The degenerate primers sequences for the dpol gene were: forward 5'-AYRYIAAYMGIGTICA IATGC-3' and reverse 5'-SIGAYCCITAYWTICCICC-3' (R= A or G; Y= T or C; M= A or C; W= A or T) (5). Reaction products were cloned into pGEM-T easy plasmids (Promega) and automatically sequenced (BaseClear, the Netherlands). The sequences obtained were deposited in GenBank under numbers: AY 895150-AY895153.

Table 1. PCR primer sequences
Other baculovirus polh, lef-8, pif-2 and dpol sequences were downloaded from GenBank to be compared with OpSNPV. The BLAST program (2) at the National Center for Biotechnology Information (NCBI) was used for nucleotide and predicted amino acid sequence homology searches. Multiple sequence alignments were done employing ClustalX. Phylogenies were constructed using Neighbouroining method of Mega 3.1. (14), using p-distance method of amino acid substitution. Gaps were treated as missing data. Tree topologies were evaluated by bootstrap analysis with 1000 replicates. The analysis of lef-8 and pif-2 was combined and the polyhedrin and dpol genes were analyzed separately. The selected genes have previously been indicated as most suitable for phylogenetic analyses (5). Additionally, lef-8 and pif-2 were found to be congruent allowing combined analysis (6). For polh gene analysis all completely sequenced lepidopteran NPVs were included as well as those showing the highest amino acid identity with OpSNPV polh gene. For lef-8/pif-2 analysis all completely sequenced lepidopteran NPVs were included (GenBank June 2007). For dpol sequence analysis, all known baculovirus DNA polymerase gene sequences that aligned with the OpSNPV dpol sequence were included. For each gene analysis an outgroup was chosen, Plutella xylostella GV polh, Xestia c-nigrum GV lef-8/pif-2 and Culex nigripalpus NPV and human DNA polymerase (dpol) genes, respectively.
-
Polh, combined lef-8 and pif-2, and dpol phylogenies clearly place OpSNPV among group Ⅱ NPVs (Fig. 1.A, B and C). OpSNPV is most closely related to two other lymantrid baculoviruses, from O. anartoides SNPV (OranNPV) and O. ericea SNPV (OrerNPV) (Fig. 1.A, and C). Analyses further show that OpSNPV is only distantly related to OpMNPV, although these two are often found intermixed in insect populations. OpMNPV is a group Ⅰ NPV (26). OpSNPV branched together with Buzura suppressaria NPV (polh and dpol trees), Ectropis obliqua NPV (dpol tree) and Clanis bilineata NPV (dpol tree). Polh sequence analysis branches OpSNPV additionally with Apochemia cinerarium NPV, but only the polyhedrin sequence is available for the latter virus and further analysis of the other genes is necessary to confirm the common ancestry of these two baculovirus species. Interestingly, polyhedrin and DNA polymerase analyses show different branching for OpSNPV and EcobNPV, suggesting that, despite distant correlation between these two species they may have acquired their DNA polymerase gene from a common source. The combined analysis of lef-8/pif-2 genes which includes only sequences of completely sequenced lepidopteran NPVs confirms the placement of OpSNPV in group Ⅱ NPVs and its close relation to EcobNPV and ClbiNPV. In this analysis however OpSNPV is placed together with LdMNPV in a common branch and this is in contrast to two other analyses. Close relationship between some open reading frames of OpSNPV and LdMNPV is not surprising as both viruses infect Lymantriidae, enabling the exchange of genetic material.
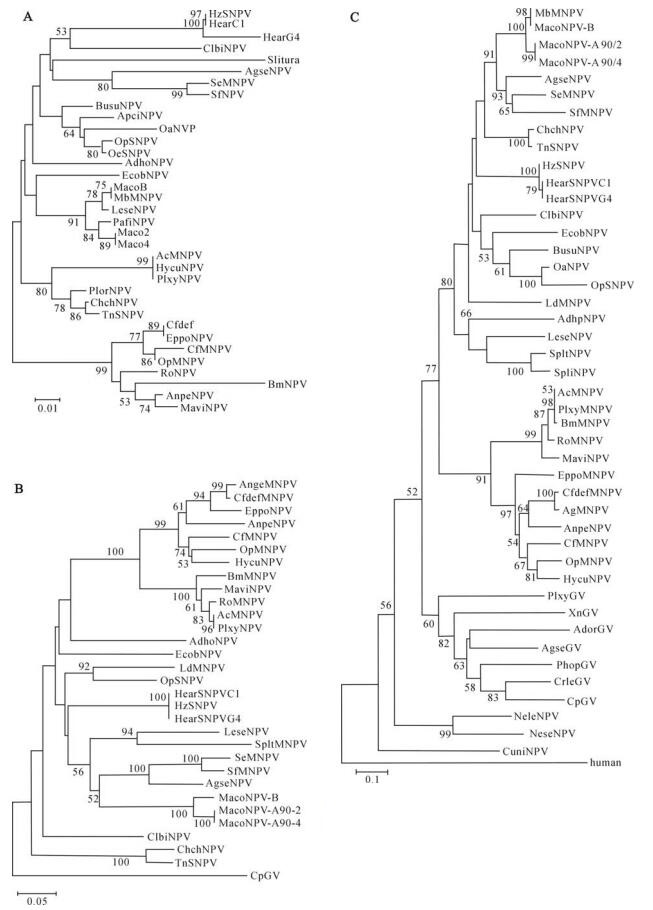
Figure 1. Baculovirus phylogenies. A: polyhedrin tree. B: Combined lef-8 and pif-2 tree. C: DNA polymerase tree. The phylogeny trees were obtained by maximum parsimony analyses of amino acids sequence data, numbers indicate bootstrap scores. Virus sequences NCBI accession numbers are collected in the Table 2.

Table 2. Baculovirus sequences used for phylogenetic analyses
-
Similarity between all known Orgyia NPVs has been implied by Hughes (8) and supported by Richards et al. (23) on the basis of biological characters. Both SNPVs and MNPVs isolated from the Orgyia genus show a high degree of cross infectivity among insect species in this genus. Their restriction profiles, however, showed enough differences to warrant a distinct taxonomic status of these two viruses (23). From the Orgyia NPVs only OpMNPV belongs to group Ⅰ NPVs. OpMNPV, which is found only in North America is closely related to another baculovirus infecting the Lymantrid, L. salicis (LeseNPV), which was described from Europe (32). Both viruses are probably derived from a very recent ancestor (11). It can even be concluded that they are variants of the same virus type and speciated recently through regional separation into separate ecological niches. OpSNPV has previously been found to share relatively similar restriction analysis profiles with O. antiqua SNPV and cross infections between both host species were documented (23). It has also been suggested that OpSNPV and O. leucostigma (Orle) SNPV are variants of the same virus infecting both species 8. Walsh et al. (28) showed that the molecular characteristics of OpSNPV are distinct from OrleSNPV and classified OpSNPV together with OpMNPV as group Ⅰ NPVs, based on their polyhedrin amino acid sequences. This finding contrasted with the study of Zanotto et al. (30), which positioned OpSNPV among group Ⅱ NPVs according to its polh gene sequence. The study of Zanotto et al. (30) indicated that the OpSNPV polh gene had an unstable position in the phylogenetic tree when its promoter sequences were included. The OpSNPV polyhedrin promoter contains elements common to the AcMNPV polh gene, the type species of group Ⅰ NPVs (not shown). Our study, based on four conserved gene sequences, clearly supports the positioning of OpSNPV within NPV group Ⅱ.
Recently, another Orgyia NPV has been isolated from O. ericea (29). This SNPV is closely related to OpSNPV on the basis of the polyhedrin sequence (Fig. 1A), even closer than to O. antiqua SNPV. The O. oricea SNPV is found in China and it would be interesting to study the cross infectivity of these Orgyia NPVs in O. oricea.
Molecular and biological information about baculoviruses is essential for understanding the relationships within this family of viruses and to promote the use of baculoviruses in insect pest control strategies. Knowledge of the taxonomic position of baculoviruses and their host range is essential in bioinsecticides registration procedures. Most baculovirus phylogenies so far are based on a single gene, usually the polh gene as it is available for a large number of baculoviruses. The polh tree topologies do not always match with tree topologies of concatenated sequences and, although very useful, need complementation with other genes phylogenies. Here we report that the OpSNPV polh gene phylogeny, positioning this virus in group Ⅱ NPVs, is supported by the analysis of sequences of three other genes, lef-8, pif-2 and dpol, which are conserved in all baculoviruses to date. Finally, this paper shows that it is highly recommended to use a universal set of primers for four conserved genes to generate reliable and robust information for the classification of baculoviruses.

 DownLoad:
DownLoad: