











-
Since the discovery of Human immunodeficiency virus (HIV) and acquired immunodeficiency syndrome (AIDS) in 1981 (50), this disease has become one of the most significant public health challenges globally. HIV/AIDS have claimed the lives of more than 25 million people worldwide. The year 2006 marks the 10th anniversity of the introduction of highly active antiretroviral therapy (HAART), a combination of three antiretrovirals (ARVs) from at least two drug classes (27, 28), that has led to significant reductions in HIV-related morbility and mortality (6, 52, 56). However, HIV virus is capable of mutating rapidly to develop drug resistance, so it is imperative to develop more effective and safe drugs to overcome the growing resistance of the virus. New molecules may block the known viral targets or other new ones. Currently there are 23 FDA-approved individual antiretroviral drugs that are classified into four categories based on their mechanism of action. There are 7 combinations of drugs currently approved by FDA. Additional agents are in various stages of clinical and preclinical development.
HTML
-
HIV-1 is an enveloped virus that contains two copies of viral genomic RNA in its core. The approximately 9 kb RNA genome encodes at least 9 proteins: Gag, Pol, Env, Tat, Rev, Nef, Vif, Vpu and Vpr, of which only the former five are essential for viral replication in vitro. The infection begins with the attachment of the virions to the cell surface mediated by an interaction among the extracellular domain of HIV-1 gp120, cellular receptors CD4 (51, 73) and chemokine coreceptors CCR5 and CXCR4 (12). After binding to coreceptor, viral and cellular membranes fuse and the viral core is released into the cytoplasm of the cell. The viral RNA genome is retrotranscribed into a full-length double-stranded DNA by the viral reverse transcriptase (RT) (30). Linear doublestranded DNA in the preintegration complex inserts into the host chromosome by the viral integrase (7, 63, 70). Expression of viral genes leads to production of precursor viral proteins. These proteins and viral RNA are assembled at the cell surface into new viral particles and leave the host cell by budding (19, 53, 55). Budding triggers the activation of the protease (PR) that autocatalytically cleaves the Gag and Gag-Pol polyprotein releasing the structural proteins and enzymes. The individual proteins undergo further interactions, with capsid and nucleocapsid protein forming the conic nucleocapsid, and matrix protein remaining associated to the viral envelope (16, 37, 75).
-
HIV entry is a critical event of HIV-1 life cycle. It is triggered by gp120 attachment to CD4, followed by gp120 engagement with a co-receptor (either CCR5 or CXCR4). Binding of HIV to co-receptors causes conformational changes in the envelope proteins, ultimately resulting in the fusion of the viral envelope and the host cytoplasmic membrane (5). As the earliest event, HIV entry represents an attractive target. In the past, this entry process between virus and host cell or transmission between cell and cell have been reestablished in vitro through using virus with reporter acceptor cell, reporter virus with reporter cells, in-fected cell and reporter cell or two reporter cell (74). But in recently years, more methods targeting one step of entry have been developed. Now, there are three types of inhibitors including: attachment inhibitors, co-receptor binding inhibitors and fusion inhibitors. Using soluble recombinant gp120 and recombinant CD4 can mock the attachment of virus to CD4+ cells in vitro, and an ELISA was developed (72). When synthetic mimetic peptides of the CD4-binding site of HIV-1 gp120 or the gp120-binding site of CD4 were used, this method was improved and new methods (such as fluorescence polarization as shown in Fig. 1) were developed with more convenience (18, 67). As one of hottest targets for entry, the binding of gp120-gp41 to co-receptor can be blocked by che-mokine antagonists. In early days, a cell-based ELISA assay using radio-labeled chemokine was widely used to measure the inhibitory activity of compounds (2). But now more other convenient methods are also used, such as baculovirus based assay and flowcytometry based assay or ELISA-based assay (33, 79). As a hottest target for entry, the process of rearrangement of gp41 to form the six-helix bundle has been investigated more intensively. Liu et al reestablished the six-helix bundles using C34 and N36 peptides in vitro (43). Based on that, many methods such as Sandwich ELISA, FLISA and FRET were developed to target this process and used in entry inhibitor screening with more convenience (76, 40).
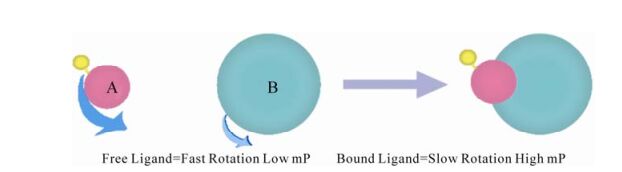
Figure 1. Fluorescence polarization in screening inhibitors of gp120-CD4 binding and protease (18, 67, 62). In screening inhibitors of binding, A and B represent fluorescein-labelled CD4 peptide and mocked CD4 binding site of HIV-1 glycoproteins respectively. In screening inhibitors of protease, A and B represent fluorescein and antigen labelled peptide substrate and antidody respectively.
-
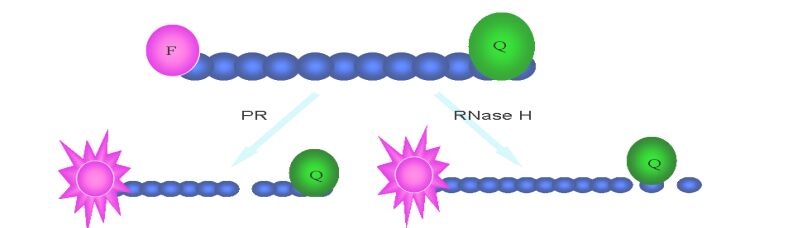
Reverse transcription is an important event of HIV life cycle. It was catalyzed by reverse transcriptase, which was encoded by HIV pol gene and consists of a 66 kDa polypeptide and a 51 kDa polypeptide with separate DNA polymerase and ribonuclease H (RNase H) domains in the 66 kDa polypeptide. Both the DNA polymerase and RNase H activities of RT are required for viral replication. In the infected cell, HIV-1 RT ultimately transcribes a single-stranded viral RNA template into double-stranded DNA (dsDNA) through a multi-step process: (ⅰ) RNA-dependent DNA polymerization to produce a (−) DNA copy, with (ⅱ) concomitant cleavage of the RNA strand of the heteroduplex by RNase H, and (ⅲ) DNA-dependent DNA polymerization to yield the dsDNA product. During this process, template switching by the newly synthesized (−) DNA takes place at least twice (21). RT inhibitors are the first HIV-1 drugs marketed and currently serve as the backbone of most frontline HIV combination therapy. Now there are three kinds of assays available for HIV inhibitor screening. First, assays for measuring DNA polymerase activity, such as Scintillation Proximity Assay(SPA)(Fig. 2), Microarray Compound Screening technology (µARCS) and enzyme immunoassays (EIAs) (38, 77, 78). In µARCS, the nucleic acid substrate was biotinylated on one end and was bound to the SAM membrane. A low melting-point agarose gel containing the rest of the reaction components was first placed on a polystyrene sheet spotted with compounds to allow passive diffusion of the compounds into the gel. The gel was removed from the compound sheet and applied to the SAM membrane with the immobilized nucleic acid template to initiate the polymerization. After the incubation, the membrane was washed with a high-salt buffer and exposed for imaging (77). Second, assays for measuring RNase H activity. In 1996, Rychetsky first development an EIA using a duplex substrate labeled with biotin and digoxigenin to high throughput screen inhibitor of RNase H (61). Recently, this method was development as a FRET assay that labels the duplex with fluorescein and Dabcyl H (Fig. 3) (57). Third, assays for measuring polymerase activity and RNase activity simultaneously. In this kind of assay, SPA assay was also used widely (20).
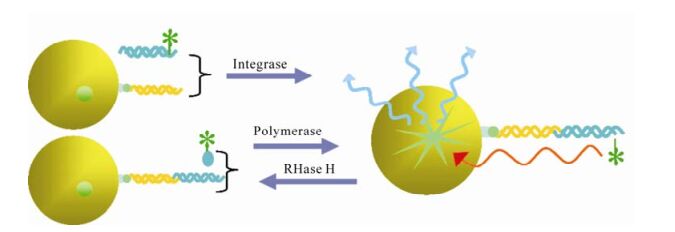
Figure 2. Scintillation Proximity Assay (SPA) in screening inhibitors of polymerase, RNase and integrase (5, 20, 38). When certain radioactive atoms decay, they release β-particles. The distance these particles travel through an aqueous solution is dependent on the energy of the particle. If a radioactive molecule is held in close enough proximity to a SPA Scintillation Bead or a SPA Imaging Bead, the decay particles stimulate the scintillant within the bead to emit light, which is then detected in a PMT-based scintillation counter or on a CCD-based imager respectively. However, if a radioactive molecule is free in a solution containing SPA beads, the decay particles will not have sufficient energy to reach the bead and no light will be emitted. This discrimination of binding by proximity means that no separation of bound and free ligand is required.
-
HIV protease is a 22 kDa homodimer aspartyl protease, which can cleave HIV Gag and Pol polypeptide precursors to form mature structural and enzymatic gene products. HIV-1 PR activity is critical for viral replication, and so it is also an ideal target for HIV therapy. In the past years, HPLC analysis of production of cut synthetic peptide substrate was used widely in protease inhibitor identification (31). But this method is too time and laborious-consuming to screen large number of samples. Subsequently, a colorimetric assay using unlabeled peptide substrate was also used with less time-cost but lower sensitivity (65). Nowadays, one synthetic peptide substrates typically consisting of a cleavage sequence flanked with fluorescent donor and acceptor labels was universally used, and the method was developed as FRET (Fig. 3). Although EDANSC/DABCYL pair is most commonly donor/acceptor labeling the peptide substrate in FRET assay, there are also other pairs with more advantage than them, such as Hilyte fluor 488/QXL 520, Hilyte Fluor TR/Qxl 620 and Cy3/ Cy5Q (22). There are also other fluorescence and label technology, such as homogeneous time-resolved fluorescence (HTRF) assays (Fig. 3), alphascreen (Fig. 4) (Perkin Elmer, Alphascreen Technical Manual) and fluorescence polarization (Fig. 1) (35, 62). In addition to enzyme assays in vitro, a number of cell-based assays have been reported for HIV-1 protease. In these assays, HIV PR expression plasmid is transfected into the reporter cell or cotransfected with plasmid in-cluding reporter gene, and then repoter gene is induced to express. Ultimately, through measuring cell viability and expression level of reporter gene, the activity of the compound is evaluated (10, 39).

Figure 4. Alphascreen assay in Screening inhibitors of protease (35).
-
HIV IN is a 32kDa dimmer protein encoded by 3'-end of pol gene. It comprises three structural domains: the aminal terminal domain (NTD) with HHCC motif, catalyze core domain (CCD) and carboxy terminal domain (CTD) with overall SH3 fold and nonspecifical binding activity to DNA required for process of integration. Through two consecutive steps: 3'-processing and strand transferring, it can insert the viral cDNA ends into host chromosomes. Integrase is essential for retroviral replication, and the absence of a host-cell quivalent of integrase means that the integrase inhibitors do not interfere with normal cellular processes, and therefore have a high therapeutic index (60). The screening and discovery of integrase inhibitors generally relies primarily on simple assays that use recombinant integrase and short oligonucleotide substrates that mimic the viral DNA ends. At the beginning of the screen, EIA is used commonly. It immobilized the donor dsDNA onto microplate, and then enzyme and target dsDNA being labeled were added, followed by ligated production quantified with the method compared to label (13, 32). With the same protocol, a more high throughput method was developed with donor dsDNA immobi-lized on membranes and targets DNA labeled with flurescein (15). Using this method, a library of 250, 000 compounds was screened for IN activity. In 2004 a method more compatible with HTS using robotics was established. Different from EIA, in SPA assay (Fig. 2) the median immobilized donor DNA was replaced by SPA beads, and the label was changed to isotope compared with the measurement method of scintillation counting (5). Besides recombinant enzyme based assays, there is a more authentic assay mimicing the reaction in vivo. The double-strand DNA targets are immobilized on 96-well plate, and preintegration complex (PIC) containing host and virus protein is added. After reaction is completed, the products are quantified by PCR (29, 49).
-
It has been proved that the formation of homodimer, pseudohomodimers or multimers play a central role in function of HIV RT, PR and IN and disruption of protein-protein interactions in enzymes may constitute an alternative way to achieve HIV-1 inhibition (8). Now a hybrid assay and an affinity assay were used in identifying inhibitors of HIV RT dimerization and many inhibitors have been found (23, 64). For in-tegrase and protease, only a few compounds were found with activity against formation of dimmer or multimer, but there aren't valuable assays targeting these two proteins.
-
In addition to the viral structural proteins (Gag and Env), and the pol-encoded enzymes (PR, RT, and IN), the HIV genome encodes several additional structural proteins (NC, MA), regulatory proteins (Rev and Tat) and accessory proteins, such as Vif, Vpu, Vpr, Vpx, and Nef. More recent tissue culture and in vivo experiments have revealed a strong requirement for these gene products for efficient virus replication and disease induction (17). And many assays have been developed targeting these proteins (Table 1). Using such assays more drugs with mechanisms different from recent approved drugs may be found in future.

Table 1. Other targets used for anti-HIV/AIDS drug screening
HIV entry
Reverse transcriptase
HIV protease
HIV integrase
Dimerization or multimerization of HIV RT, PR and IN
Other targets
-
DC-SIGN, a lectin expressed on dendritic cell and macrophage subsets, binds to human immunodeficiency virus Env glycoproteins, allowing capture of viral particles. Captured virions either infect target cells or are efficiently transmitted to lymphocytes (46). It was found that competition between CD4, bovine lacto-ferrin or lactoferrin and DC-SIGN for Env binding likely can affect virus access to the cytosol, syncytium formation and prevent dendritic cellmediated human immunodeficiency virus type 1 transmission (26, 54). A group of carbohydratebinding agents (CBAs) also have this effect (3). These findings indicate that it is a potential target for anti-HIV therapy, but the side-effects resulted from blocking this receptor haven't been clarified and this may obstruct inhibitor deve-lopment.
-
Lens epithelium-derived growth factor/transcription co-activator p75 (LEDGF/p75) protein is an essential HIV integration cofactor (42). It forms a specific nuclear complex with integrase through interaction with both the N-terminal zinc binding domain and the central core domains of IN. And this interaction was conserved within and limited to lentivirus and is intimately involved in the catalysis of lentiviral DNA integration (11). This interaction with LEDGF/p75 accounts for the karyophilic properties, chromosomal targeting of HIV-1 IN, protection of HIV-1 IN from the proteasome (41, 44). The data from competition of the IBD fusion proteins with endogenous LEDGF/p75 for binding to integrase provide proof of concept for the LEDGF/p75-integrase interaction as a novel target for treating HIV-1 infection (71).
-
Conformational changes within the human immu-nodeficiency virus-1 (HIV-1) surface glycoprotein gp120 result from binding to the lymphocyte surface receptors and trigger gp41-mediated virus/cell me-mbrane fusion. The triggering of fusion requires cleavage of two of the nine disulfide bonds of gp120 by a cell-surface protein disulfide-isomerase (PDI). Using surface plasmon resonance, Barbouche et al found heparin and heparan sulfate can reduce PDI-mediated gp120 reduction by approximately 80%. And it was also found that interaction of Env with the surface of lymphocytes being treated with sodium chlorate, an inhibitor of glycosaminoglycan synthesis, led to reduction of gp120 (4). Gallina et al reported that monoclonal antibodies to protein-disulfide isom-erase (PDI) and other membrane-impermeant PDI inhibitors prevented HIV-1 infection (21). Markovic also found this phenomenon with other anti-PDI agents (45). And exogenously added PDI, in turn, can rescue fusion from this blockade (45). All these events described provide clues for identifying new potential targets for screening of new anti-HIV agents.
-
Proteasomes constitute the degradative machinery of the ubiquitin/adenosine triphosphate-dependent proteolytic pathway, which is involved in many cell functions, including immune response and apoptosis (59). Recent findings showed that proteasome inhibitors are described to interfere with HIV matu-ration, budding and aggressiveness, and cytostatic drugs, as well as antiretroviral agents used in HAART, have been shown to behave in vitro and in cultured cell lines as inhibitors of proteasome proteolytic activity at therapeutic dosages (58). In investigation of the function of the ubiquitin-proteasome system in HIV replication, Gottwein et al observed that, like several other retroviruses, HIV-1 virions contain a small amount of mono-ubiquitinylated Gag proteins (24). In another analogous investigation, Amit et al found that, in addition to one E3-like protein, two E3-type ubiquitin ligases also could act as regulators of HIV budding (1). These ligases might represent interesting targets for therapeutic intervention (36).
Interaction of DC-SIGN and gp120
Interaction of LEDGF/p75 and integrase
Disulfide-isomerase (PDI)
Ligases proteasome system
-
Although the future of antiretroviral therapy is challenged by side effects and drug resistance, there are prospects for novel drugs. The emerging understan-ding of viral replication may enable the development of new therapeutic targets. With more new assays and small molecule synthesis technology available, more potent new anti-HIV drugs are to be developed and used in the treatment of AIDS.

 DownLoad:
DownLoad: