











-
Influenza A viruses belong to the family Orthomyxoviridae, and currently they are classified into 16 haemagglutinin (HA) and 9 neuraminidase (NA) subtypes[6, 33]. Generally, avian influenza viruses cannot efficiently infect humans despite sporadic human infection events involving avian H5N1 viruses in recent years[18]. Pigs, however, are susceptible to both avian and human viruses because their tracheal epithelium possesses both sialic acid (SA)-a-2, 6-Gal-terminated saccharides and SA-a-2, 3-Gal receptors[11], and have been postulated to serve as unique candidates for the generation of human, avian, and/or swine reassortant viruses with the potential to cause a pandemic in humans, which is exemplified by A(H1N1) pdm09 viruses through reassortment of swine-origin viruses[24].
Influenza A virus infection is an important febrile respiratory disease in pigs[3]. Currently, three predominant subtypes of influenza virus are prevalent in pig populations worldwide: H1N1, H3N2, and H1N2[3, 23, 31]. The antigenic and genetic diversity of swine H1N1 viruses have been documented in different parts of the world. The classical swine virus (H1N1) was evolved from the 1918 pandemic, and first isolated in 1930. Since then its progeny viruses continued to circulate in swine populations in Asia, North America and until 1979 in Europe[3, 32]. Since 1979, the previously dominant classical H1N1 swine viruses have been replaced by the avian-like H1N1 viruses which antigenically and genetically are closely related to Eurasia avian H1N1 viruses[3, 19]. These two distinct lineages of swine H1N1 viruses (classical and avian-like) showed different evolutionary trajectories during swine adaptation[4]. Since 1984, reassortant H3N2 and H1N2 viruses containing human HA and NA and avian-like internal genes (PB2, PB1, PA, NP, M and NS) have circulated in pigs in Europe[3]. In North America, classical swine H1N1 viruses predominantly circulated in pigs before 1998, and triple-reassortant swine (TRS) H1N1, H1N2, and H3N2 viruses containing genes of human, swine and avian viruses have become established in pigs since this time[3, 12, 17, 32, 38].
On 7 January 2011, a case with a 3-year-old boy was infected with avian-like H1N1 swine influenza virus was confirmed in our lab by virus isolation and sequencing. The subject lived in a village where farmers often raise animals in their courtyards, such as pigs, ducks and chicken. In order to identify the genetic origin of the isolates, in this study, we performed molecular characterization of two influenza A viruses isolated from pigs collected from the villages neighboring where the subject lived. The results indicated that two isolates were closely related in all eight genes to European avian-like swine H1N1 viruses, especially those circulating in pigs in China. Furthermore, the two swine isolates and the human isolate probably derived from the same progenitor viruses.
HTML
-
From January to April 2011, we obtained 60 lung samples from slaughtered pigs collected from the neighboring villages to where a 3-year-old boy infected with avian-like H1N1 swine influenza virus had lived. The lung samples were ground, and 10 % suspensions with Hanks` Balanced Salt Solution were made. 200 μL of each processed specimen was inoculated into Madin-Darby canine kidney (MDCK) cells, which were maintained in Dulbecco's Modified Eagle Medium (DMEM, Gibco) with TPCK-trypsin (2 μg/mL). The viruses were harvested when 75 %-100 % cytopathic effects were observed, and stored at -70 ℃. The isolates were typed by hemagglutinin inhibition (HI) and neuraminidase inhibition (NI) assays and genome sequencing[21].
-
Viral RNAs were extracted from 140 μL lung samples with an RNeasy Mini Kit (Qiagen). PCR was performed with a set of primers specific for each gene segment of influenza A virus[10]. PCR products were purified by using a QIAamp Gel extraction kit (Qiagen) and sequenced using an ABI 3730 DNA Analyzer (Applied Biosystems). The nucleotide BLASTn analysis was used to identify related reference viruses, and the reference sequences were obtained from GenBank. More than 60 reference viruses were selected, comprising H1N1 viruses from poultry, human and swine. Phylogenetic analysis of 8 gene segments was performed by the Maximum Composite Likelihood model in software MEGA 4.1[28]. The reliability of the unrooted neighbor-joining tree was assessed by bootstrap analysis with 1000 replications. Horizontal distances are proportional to genetic distance. Alignments of each influenza virus sequence were created using ClustalX 1.83 (www.clustal.org/).
Virus isolation and identification
Genomic sequencing and phylogenetic analysis
-
Six viruses were isolated from 60 lungs samples, and HI and NI assay showed they belonged to the European avian-like swine H1N1 subtype, which were confirmed by genomic sequencing and BLASTn analysis. Two isolates, A/swine/ Jiangsu/s15/11/2011 (s15/11) and A/swine/Jiangsu/s16/11/2011 (s16/11), were selected to represent all six viruses and subjected to further genetic analysis. A/Jiangsu/ALS1/2011 (ASL1/11) was isolated from a 3-year-old boy admitted to hospital with severe pneumonia on 3 January 2011, who subsequently was confirmed to be infected with European avian-like H1N1 swine influenza virus in our lab (data not shown).
-
The complete eight gene segments of s15/11 and s16/11 were sequenced, and the nucleotide sequences have been deposited in the GenBank database under accession numbers JF820274-JF820289. The genetic origin of the two isolates were initially determined by a BLAST search against GenBank and pairwise comparisons of each gene segment to the corresponding sequences of reference viruses. The nucleotide sequence identities among the eight genes of the two isolates were 99.7 % (NP, PB1)-100 % (NA, PB2, PA), which suggests that the two isolates come from the same progenitor viruses.
The eight genes of the two isolates shared the highest nucleic acid sequence similarity with those of European avian-like H1N1 isolates circulating in China since 2006 (98.1 %-99.8 %) (Table 1) and amino acid sequence similarity ranged from 97.5 % (NEP) to 100 % (M1 and M2) (data not shown). They were clearly distinct from the corresponding genes of recent classical swine H1N1, human seasonal H1N1, A (H1N1) pdm09, and avian H1N1 viruses, represented by A/swine/Shanghai/1/2005 (Sw/SH/1/05), A/Brisbane/59/2007 (A/Brisbane/59/07) and A/ Duck/Italy/69238/2007 (Dk/Italy/69238/07), respectively (Table 2). The NA and M genes of the two isolates are less closely related to those of reference strain A/Jiangsu/1/2009 (A/JS/1/2009) (90.0 % and 95.3 %-95.5 % similarity, respectively), a A(H1N1) pdm09 virus with the NA and M gene derived from European avian-like swine H1N1 virus which was isolated in our lab in 2009. In contrast, relatively low sequence similarities ( < 84.3 %) were found with the other 6 genes (HA, NP, NS, PB2, PB1 and PA genes) between the two isolates and A/JS/1/2009. To determine more precisely the genetic origin of the two isolates, we constructed phylogenetic trees, including reference viruses consisting of H1N1 viruses isolated in poultry, human and swine (Fig. 1). In the HA tree, there are seven distinct lineages comprising seasonal human H1N1, classical swine H1N1, A(H1N1) pdm09, northern American triple re-assortment H1, Eurasian avian H1N1, American avian H1N1, and European avian-like swine H1N1 viruses. The two isolates clustered with European avian-like swine H1N1 viruses, and were located within a sub-lineage comprising swine viruses isolated in China from 2006 to 2010. The seven lineages were also present in the phylogenic trees of the other seven genes (NA, NP, M, NS, PB2, PB1 and PA), and the two isolates grouped with European avian-like swine lineage in each of the seven trees. These results further confirmed that each of eight genes of the two isolates were closely related to the avian-like H1N1 viruses circulating in pig populations, with very high similarity to those isolated in China.

Table 1. Nucleotide sequence identity between A/swine/Jiangsu/s15/2011 and A/swine/Jiangsu/s16/2011 and reference strains downloaded from GenBank

Table 2. Amino acid residues associated with receptor-binding sites of HA proteins of A/swine/Jiangsu/s15/2011 and A/swine/ Jiangsu/s16/2011 and reference viruses
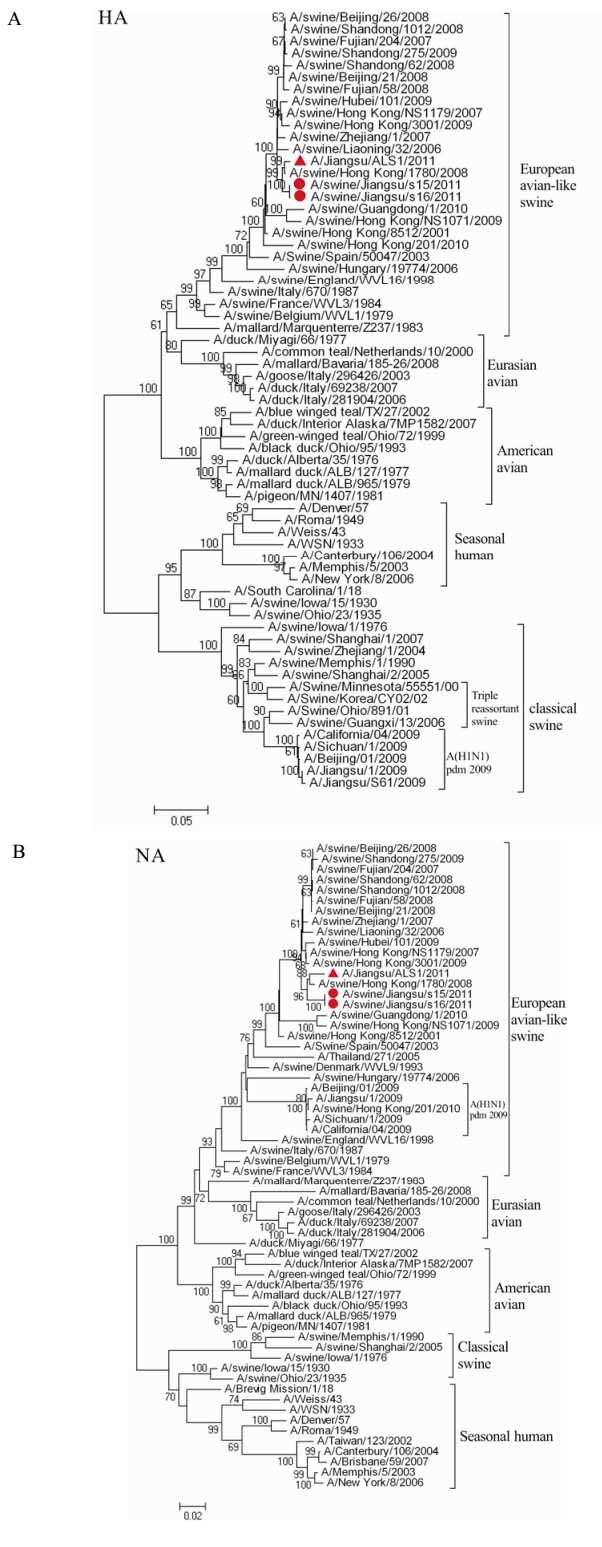
Figure 1. Phylogenetic trees for the HA (A) and NA (B) genes of A/swine/Jiangsu/s15/11/2011 and A/swine/Jiangsu/s16/11/2011 and related reference viruses. The unrooted neighbor-joining phylogenetic trees were generated by the Maximum Composite Likelihood model in software MEGA 4.1 (www.megasoftware.net). The reliability of the tree is assessed by bootstrap analysis with 1, 000 replications. Bootstrap values are shown for selected nodes (only for those with a frequency greater than 70 %) Horizontal distances are proportional to genetic distance. The sequence regions compared were as follows: HA (1699bp), NA (1382 bp). The isolates in this study are marked with a circle. Reference strain A/Jiangsu/1/2009 is marked with a triangle.
-
Based on the deduced amino acid sequence, the isolates were found to contain an amino acid motif PSIQSR↓G at their HA cleavage sites, a characteristic of low pathogenic influenza viruses[13]. There are four potential glycosylation sites (Asn-X-Ser/Thr), observed at positions 13, 26, 198, 277 in the HA1 proteins of the two isolates. The two isolates contain the following amino acids at residues 71D, 138A, 155V, 159N, 186S, 190D, 194L, and 225E (according to the H3 site numbering), which were previously identified as receptor-binding sites (Table 2). Compared with human isolate ALS1/11, the s15/11 and S16/11 isolates had four amino acid mutations (N87D, L210F, R267M and R354Q) in the HA protein (98.5 %-98.6 % nucleic acid sequence similarity, 99.3 % amino acid sequence similarity), all of which were not located in previously defined antigenic sites or receptor-binding sites[16, 34].
The amino acid substitutions (H274Y and N294S) were not observed in the NA proteins of the two isolates, which suggests that they are sensitive to oseltamivir and zanamivir. Also, the M2 proteins of the two isolates have the mutations S31N, which could confer resistance to amantadine and rimantadine antivirals, a characteristic marker of the European avian-like swine viruses (H1N1, H3N2 and H1N2) since about 1987[8, 9, 14, 26, 27, 36].
Based on the 1918 pandemic H1N1 and highly pathogenic avian H5N1 viruses, many determinants of virulence and host adaptation have been identified[1, 13]. Due to the presence of a stop codon at codon 12, both isolates contained a truncated PB1-F2 protein. The amino acids at 591, 627 and 701 in PB2 have been shown to be determinants of virulence and host adaptation[9, 26, 27, 36]. In this study, the isolates included 591Q, 627E and 701N within the polymerase subunit PB2. The isolates also had D rather than E at position 92 of the NS1, a marker of mammalian adaptation[1]. The two isolates contained the GPKV motif at the PDZ ligand domain of the 3' end of the NS1, a characteristic marker of the European avian-like swine viruses since about 1999[31], which was distinct from those of avian, human and classical swine viruses.
Virus isolation and identification
Phylogenetic analysis
Molecular characterization
-
In this study we investigated the molecular characteristics of two strains isolated from swine lung samples collected from January to April 2011 in eastern China. Our results showed that isolates s15/11 and s16/11 were closely related to avian-like swine H1N1 viruses recently circulating in pigs in China, which were initially detected in European pigs in 1979. Phylogenetic analyses results indicated that genetic re-assortment with human or avian viruses had not occurred. In 60 swine lung samples, no other genotype influenza viruses, such as the classical swine, triple re-assortment viruses and A (H1N1) pdm09-like viruses, were detected. The results provided further evidence that European avian-like H1N1 viruses have been circulating in pigs in eastern China since 2007[15].
The molecular mechanism of virulence and host adaptation ofinfluenza A viruses remains unclear and may involve both multiple viral genes and host factors[18, 33]. The receptor binding specificity of HA has been implicated as a major determinant of the host range of a given influenza virus. Some studies revealed that two amino acid mutations of H1 HA, E190D and G225D/E, could cause a shift in receptor binding specificity from the avian SA-a-2, 3-Gal to the human SA-a-2, 6-Gal[7, 16, 29]. In this study, the two isolates contained 190D and 225E, which may imply the viruses have the same receptor binding preference as human viruses. Two amino acid mutations (E627K or D701N) of the PB2 protein are known to be important for adaptation of avian viruses to mammals[26, 27]. A recent study showed that a basic amino acid (such as R) at position 591 of the PB2 could increase viral replication and virulence in mammals, which might compensate for the lack of 627 K[36]. In this study, similar to avian influenza viruses, the two isolates contain 591Q and 627E in PB2, but had an N at position 701 of PB2, which may increase their fitness in mammals.
Although systematic surveillance of swine influenza viruses is lacking, early studies have indicated that virus subtypes H1N1, H1N2 and H3N2 are present in pigs in China[2, 20, 31, 37]. European avian-like swine H1N1 viruses may have appeared in 2001 in China, and gradually became dominant in pigs since then[15, 31]. North American triple-reassortment swine H1N2 viruses were also isolated in recent years[35]. Additionally, interspecies transmission of avian H5N1 and H9N2 to pigs have been reported in China[31]. These findings demonstrate that genetic evolution of influenza viruses is complex and diverse in pigs in China. Some studies showed that there were correlation between genetic re-assortment and antigenic evolution[22, 31, 32]. Reassortment events frequently occur in pigs, which led to the genetic diversity of influenza viruses in swine population. Though most of novel re-assortants disappeared, a few of them have become established. Following the generation of the A (H1N1) pdm09 viruses, pdm09-like and reassortant viruses containing genes of pdm09-like and other influenza viruses have frequently been detected in pigs from different countries, including China[25, 30, 31, 39]. Most recently, new H3N2 reassortant viruses with pdm09 internal genes have prevailed in pigs in southern China[5]. From August to December 2011, 11 cases were reported infected with novel triple reassortant swine H3N2 viruses of swine origin containing the M gene from A(H1N1) pdm09. All these findings further emphasize the importance of surveillance for genetic diversity of influenza A viruses in pigs, and raise further concerns about the occurrence of cross-species transmission events.


 DownLoad:
DownLoad: