











-
West Nile virus (WNV), the etiologic agent responsible for West Nile encephalitis (3, 21), belongs to the genus Flavivirus. Besides WNV, many other flaviviruses also cause significant human diseases, including yellow fever virus (YFV), four-serotypes of dengue virus (DENV), Japanese encephalitis virus, and tick-borne encephalitis virus (4, 19). More than 50 million, 200 000, and 50 000 humans are infected by DENV, YFV and JEV every year, respectively (9). Flavivirus virions are about 50 nm in diameter, and harbor a plus-sense, single-stranded RNA genome of about 11 kb. The single open reading frame of the genome encodes a polyprotein, which is processed by viral and cellular proteases into three structural proteins [capsid (C), premembrane or membrane (prM/M), and envelope (E)] and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (5). Among those, the structural proteins are involved in viral particle formation. The NS proteins are primarily responsible for RNA replication; they also function in viral assembly (13, 15) and evasion of host immune response (16, 20). Serological assay is the dominant method for diagnosis of flavivirus infections in human. This is because, by the time patients develop symptoms and present to clinicians, immune response has already cleared the viruses or reduced the viremia to very low or undetectable levels. Upon flavivirus infection, majority of the neutralizing antibodies are directed against the E protein, although some antibodies may recognize the prM/M protein (6, 8, 22, 27). These antibodies are cross-reactive with other flavivirus members, leading to specificity as the confounding challenge of the serological diagnosis of flavivirus infections. The current serology assays use viral structural proteins (inactivated virus or recombinant E protein) as antigens in ELISA or Luminex formats (10, 14, 18, 26). These serology assays lack specificity, verified by cross-species plaque reduction neutralization tests (PRNT) before a specific virus of infection could be determined. PRNT is performed by testing the neutralization of a panel of flaviviruses by a potential flavivirus-infected serum (17); it represents the most accurate serologic method for differentiating infections of closely related flaviviruses. Since PRNT uses live viruses, the assay should be performed in biosafety level-3 or level-4 containment for many flaviviruses. Additionally, PRNT is time-consuming, requiring 10, 7, and 5 days to test DENV-1, YFV, and WNV, respectively (23). Here we describe a novel approach for virus-type specific diagnosis of flavivirus infection using viral-like particles (VLPs). The VLPs contains a luciferase-reporting replicon that is encapsidated and enveloped by the complete viral structural proteins. The VLPs have physiochemical and antigenic properties similar to the authentic virus particles, and can infect susceptible cells. However, the infection is a single-round, because no structural proteins are expressed upon the VLP infection. Using a WNV-infected mouse serum, we showed that the serum neutralized WNV VLP more selectively than the DENV-1 and YFV VLPs. The results demonstrate that VLPs can replace raw viruses for neutralization assays to differentiate closely related flaviviruses. Compared with the raw virus-based PRNT, the VLP-based neutralization assay is rapid ( < 1 day) and quantitative (measured by luciferase activity), and can be performed in biosafety level-2 laboratory.
HTML
-
Vero and BHK-21 cells were maintained in Dulbecco modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) at 37℃. WNV strain 3356 was used in this study. WNV-infected mouse serum (immune IgG) was a generous gift from Kristen Bernard (Wadsworth Center, Albany, New York). The anti-actin antibody was purchased from Sigma. The monoclonal antibody against WNV NS5 was in-house generated.
-
Ten micrograms of flavivirus replicon containing a Rennila luciferase and the foot-and-mouth-disease virus 2A (Rluc2A-Rep) were electroporated into 8×106 of BHK-21 cells by a Gene Pulser Xcell apparatus (Bio-Rad) at 850 V and 25 μF (23, 24). Then 4×105 and 2×105 transfected cells were seeded in a 12-well plate per well for assaying luciferase signals at 0-24 h post-transfection (p.t.) and at 24-48 h p.t., respectively. At various time points, the cells were washed once with cold PBS, and treated with 250μL of 1×lysis buffer (Promega). The plates containing the lysis buffer were sealed with Parafilm and stored at -80℃. Once samples for all time points had been collected, 20μL of cell lysis was transferred to the 96-well plates, and assayed for luciferase signals in a Turner BioSystems luminometer (Promega).
-
Ten microliters of cell lysis, collected at various time points after replicon transfection, were separated on an SDS-PAGE. The proteins in the SDS-PAGE were transferred to a Hybond-C Super nitrocellulose membrane (Amersham). The nitrocellulose membrane was blocked with 5% skim milk in PBS, incubated with primary antibody (anti-actin and anti-WNV NS5) and a secondary antibody (HRP-conjugated goat anti-mouse IgG; Jackson Immuno-Research Laboratories), and developed with chemiluminescence ECL reagents (Amersham).
-
Ten micrograms of WNV (strain 3356), YFV (17D vaccine strain), or DENV-1 (Western Pacific strain) replicon (Rluc2A-Rep) were individually electroporated into 8×106 BHK-21 cells. An alphavirus Semliki Forest virus (SFV) replicon was used to express flavivirus structural genes (SFV-CprME-Rep) (23, 24). At 24 h p.t., the cells were electroporated again with 10μg of SFV-CprME-Rep RNAs. At 24 h post the second transfection, the culture fluids were harvested, centrifuged at 415 g for 5 min at 4℃ (to remove cell debris), aliquoted, and stored at -80℃. Both flavivirus Rluc2A-Rep RNA and SFV-CprME-Rep RNA were in vitro transcribed from linearized DNAs, using a T7 and SP6 mMESSAGE mMACHINE kit (Ambion), respectively.
-
Two methods were used to quantify VLPs (24). Method one used luciferase assay to compare VLP titers of the same virus-type; method two used indirect immunofluorescence assay (IFA) to estimate the VLP titers of different virus-types. Briefly, for the luciferase assay, approximately 4×104 Vero cells were seeded per well in a 96-well plate. At 24 h post-seeding, the cells were infected with 200μL VLPs, and incubated in 5% CO2 at 37℃. At 24 h p.i., the cells were washed by 200 μL of cold PBS, treated with 20 μL lysis buffer for 20 minutes on a shaker, and then measured for luciferase signals in a Turner BioSystems luminometer (Promega). For the IFA assay, Vero cells in a four Chamber Slide (Nalge Nunc International) were infected with serial dilutions of VLP samples; IFA was performed on the infected cells at 24h p.i.; and IFA-positive cell foci were counted. The VLP titers were estimated by the number of positive IFA-positive foci, and expressed in focus-forming units (FFU)/mL. Immune mouse ascetic fluid of WNV, DENV-1 and YFV (American Type Culture Collection) and goat anti-mouse immunoglobulin G conjugated with Texas Red were used as primary and secondary antibodies, respectively.
-
BHK-21 cells were plated in 12-well plates and infected with WNV VLP at 0.5 FFU per cell. At 1 h p.i., cells were washed three times with 2 mL of PBS. At the indicated time points, total cellular RNA was isolated from infected cells using the RNAeasy kit (Qiagen). The RNA was subjected to RT-PCR (Superscript Ⅲ One-step RT-PCR system; Invitrogen) amplification using two primer sets. One primer set spans the luciferase reporter, and the other primer set amplifies the N-terminal region of viral NS5 (Table 1). The RT-PCR products were analyzed using an agarose gel electrophoresis.

Table 1. Primers used for detection of luciferase reporter and NS5
-
Heat-inactivated mouse serum was diluted with BA-1, mixed with an equal volume of WNV suspension (200 PFU), and incubated for 1 h at 37℃. The antibody-virus mixtures were incubated in duplicates with Vero cells in 6-well plates. After adsorption of viruses for 1 h at 37℃, 3 mL of a first layer agar containing 0.6% Oxoid agar, basal medium Eagle with 1% FBS, 0.02% DEAE dextran, and 0.13% NaHCO3 was added to the infected cells. Two days later, 3 mL of a second layer agar containing 1% Noble agar, basal medium Eagle with 1% FBS, 0.02% DEAE dextran, 0.13% NaHCO3, and 0.004% neutral red was added over the first layer. The plates were further incubated for 12 h before plaques were counted (23). Neutralization titers are determined as the highest serum dilution that inhibits the formation of at least 90% of the plaques as compared with mock-treated virus control.
-
Serum was heat inactivated at 56℃ for 30 minutes. Ten-fold serial dilution of serum samples was mixed with an equal volume containing 100 FFU VLPs of WNV, YFV, or DENV-1. After incubation at 37℃ for 1h, 4×104 Vero cells in a 96-well plate (seeded one day before VLP infection) were infected with the neutralized VLPs. At 22 h p.i., the 96-well plates were subjected to luciferase assay as described above.
Cells, viruses, and mouse serum
Transient replication of flavivirus replicon
Western blot analysis
Packaging of virus-like particles (VLPs)
Luciferase assay and immunofluorescence assay
Stability analysis of luciferase reporter in WNV Rluc2A-Rep VLP
PRNT
VLP-based neutralization assay
-
We have previously reported the construction of WNV Rluc2A-Rep (24, 25) and DENV-1 Rluc2A-Rep (26), in which a Renilla luciferase gene was engineered in a position where the viral structural genes were deleted (Fig. 1A). A foot-and-mouth-disease virus 2A sequence was inserted into the replicon to mediate a cleavage between the luciferase and its downstream viral polyprotein. We obtained a similar Rluc2A-Rep of YFV (11) from Richard Kuhn (Purdue University, USA). Transfection of BHK-21 cells with Rluc2A-Rep of different flaviviruses yielded two luciferase peaks: the first peak was observed during the initial 6 h p.t., and a second peak was observed after 6 h p.t. (Fig. 1B). A similar time frame of the luciferase curves was observed for all three flavivirus replicons. However, the luciferase curve derived from the DENV-1 replicon was 100-to 500-fold lower than those derived from the YFV and WNV replicons (Fig. 1B), possibly due to the slow replication nature of the DENV-1. Similar two-peak luciferase kinetics was observed upon transfection of Huh 7.5 cells with the three flavivirus Rluc2A-Rep (data not shown). These results indicate that (ⅰ) the Rluc-2A-Rep RNAs of WNV, DENV-1, and YFV are replication-competent; and (ⅱ) the two luciferase peaks represent viral translation and RNA replication.To confirm that the luciferase signal correlates with the viral replication level, we monitored the kinetics of NS5 expression in the WNV Rluc2A-Rep transfected cells. Cell lysates at 4 h, 24 h, 36 h and 48 h p.t. were quantified for NS5 expression using Western blot (Fig. 1C). The expression of NS5 became detectable at 24 h p.t., and increased significantly at 36 h and 48 h post-transfection. As a control, similar amounts of actin were detected from samples harvested at different time points. Overall, the results demonstrate that the luciferase replicon can be reliably used to monitor the replication of various flavivirus Rluc2A-Rep.
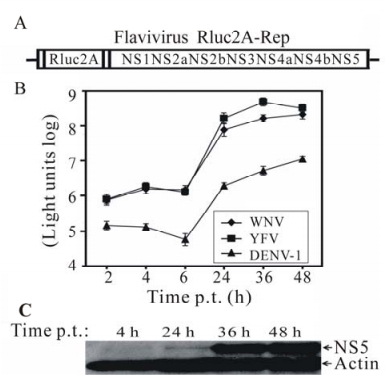
Figure 1. Flavivirus reporting replicon (Rluc2A-Rep) (A). The replicon contains a Renilla luciferase and the foot-and-mouth-disease virus (FMDV) 2A at a position where WNV structural genes were deleted. Identical replicon constructs for WNV, DENV-1, and YFV were used in this study. Transient replicon assay(B). BHK-21 cells were electroporated with 10 μg of Rluc2A-Rep of indicated flaviviruses, and assayed for luciferase activities at various time points. Expression of NS5 in WNV Rluc2A-Rep-transfected cells(C). Western blot was performed to monitor the expression of NS5 in WNV Rluc2A-Rep-transfected BHK-21 cells. Cell lysates were collected at indicated time points. Actin was used as a control for sample loading.
-
We packaged the flavivirus Rluc2A-Rep into VLPs by trans supplying homologous structural proteins. A complete structural polyprotein (CprME) of WNV, YFV, or DENV-1 was expressed through a 26S subgenomic promoter from the SFV expression vector (SFV-Flavi-CprME) (Fig. 2A). The flavivirus Rluc2A-Rep and SFV-Flavi-CprME were sequentially electroporated into BHK-21 cells; and VLPs in culture supernatants were harvested (See Materials and Methods for details). To examine the infectivity of the VLPs, we incubated naive Vero cells with the VLP supernatants. Robust luciferase activities were observed at 22 h p.i. for all three viral VLPs, in the order of WNV > YFV > DENV-1 (Fig. 2B). Next, we determined the titers of VLPs by IFA-staining of the VLP-infected Vero cells (Fig. 2C). The titers of VLPs were estimated to be 1×106, 2×105 and 2×103 FFU/mL for WNV, YFV and DENV-1, respectively (Fig. 2D). These results demonstrate that infectious VLPs can be generated by trans supplying viral Rluc2A-Rep with homologous flavivirus structural proteins.
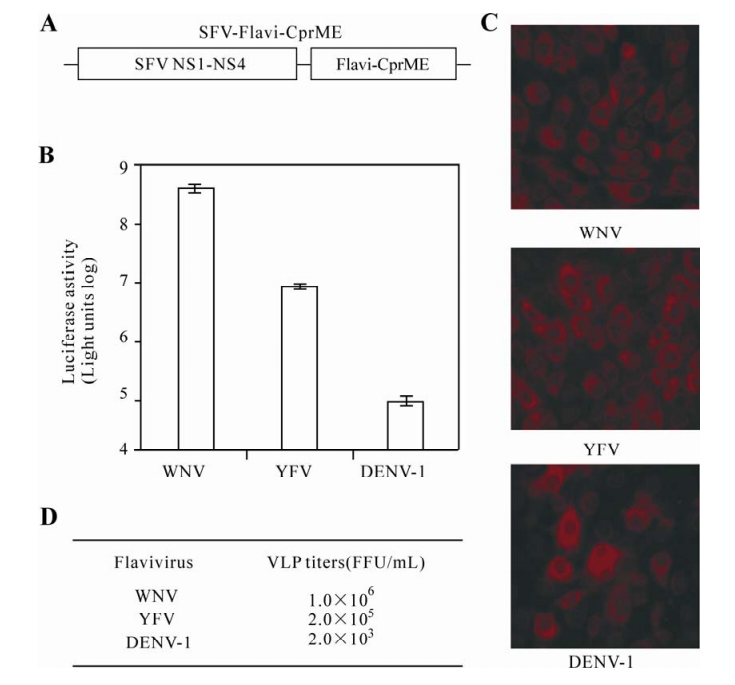
Figure 2. Homologous packaging of flavivirus VLPs. A: A complete structural polyprotein (CprME) of WNV, YFV, or DENV-1 was individually expressed through a 26S subgenomic promoter from the SFV expression vector (SFV-Flavi-CprME). B: Vero cells (4×104 per well) in 96-well plate were infected with 200 μl fluid containing WNV, YFV, or DENV-1 VLP. At 22 h p.i., the 96-well plates were subjected to luciferase assay. Averages results of four independent experiments are presented. Error bars represent standard deviations. C: Vero cells were infected with indicated flavivirus VLPs, and analyzed by IFA at 24 h p.i. (See details in Materials and Methods). (D) Summary of VLP titers. The titers were derived from IFA experiments described in C.
-
We previous showed that the luciferase gene in an infectious WNV genome was not stable. Specifically, the luciferase gene was deleted after one or two rounds of viral infection (7). This result prompted us to ask whether luciferase reporter would be deleted from the WNV Rluc2A-Rep after the preparation and infection of VLPs in cell culture. To address the question, we infected Vero cells with WNV VLP at 0.5 FFU per cell. At 48 h p.i., total cellular RNA was extracted, and subjected to RT-PCR analysis using two pairs of primers. One primer pair, WNV Aford and 2993C, flanks the luciferase gene; another primer pair, 7877V and 8804C, targets the NS5 region (Fig. 3A and Table 1). The results showed that there is no deletion in the WNV Rluc2A-rep at 48 h p.i., as compared with the control RNA transcribed from the Rluc2A-rep cDNA plasmid (Fig. 3B).
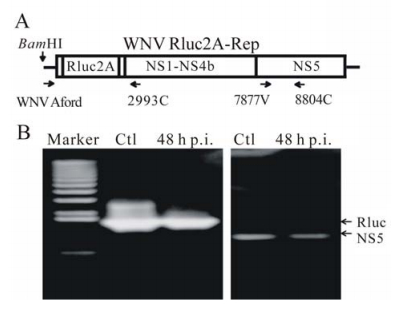
Figure 3. Stability analysis of luciferase gene in WNV VLP. BHK-21 cells were infected with WNV VLP at 0.5 FFU per cell for 1 h, washed three times with PBS, and incubated at 37℃. At 48 h p.i., total RNAs were extracted from the infected cells. The extracted RNAs were then subjected to RT-PCR amplification using two sets of primers (A): One set (WNV Aford and 2993C) flanks the Rluc2A insert, and the other set (7877V and 8804C) spans the N-terminal region of NS5. As positive controls (Ctl), the WNV Rluc2A-Rep transcript was amplified through RT-PCR. The RT-PCR reactions were resolved on an agarose gel. DNA markers and the expected bands of Rluc and NS5 region are indicated.
-
The luciferase-reporting VLPs of flavivirus are useful to study viral entry and replication. We hypothesized that VLPs could replace raw viruses in the PRNT assay, which is currently considered as the "gold standard" for serologic diagnosis of flavivirus. To test this hypothesis, we examined whether a WNV-infected mouse serum could selectively neutralize WNV VLP among a panel of flavivirus VLPs. We first used the classic PRNT to determine the titer of WNV-neutralizing antibody in the mouse serum (as detailed in Materials and Methods). The result showed that incubation of WNV with the mouse serum at a dilution of 1:160 reduced 90% of plaques. Next, we applied flavivirus VLPs to a neutralization assay. Equal amounts of VLPs (100 FFU) of WNV, DENV-1, and YFV were incubated with a serial 10-fold dilutions of the WNV-infected mouse serum (characterized above) at 37℃ for 1 h. After neutralization, the VLPs were used to infect Vero cells; the infected cells were then quantified for luciferase activities at 22 h post-infection. As shown in Fig. 4, the WNV-infected mouse serum inhibited the WNV VLP infection in a dose-dependent manner; 87%, 54%, and 16% inhibitions were observed at 103-, 104-, and 105-fold serum dilutions, respectively. For the DENV-1 VLP, 98% inhibition was detected with 10-fold serum dilution; only 13% inhibition was observed at 102-fold dilution. For the YFV VLP, only 28% inhibition was detected at 10-fold serum dilution. These results clearly demonstrate that a WNV-infected mouse serum neutralized the WNV VLP more efficiently than the DENV-1 and YFV VLPs. It should be noted that flavivirus infection generates non-infectious subviral particles, besides the complete infectious particles. These subviral particles are empty (i.e., without nucleocapsid), and can be formed when only prM and E are expressed (in the absence of viral RNA and C protein). Therefore, during the process of VLP production, a mixture of infectious VLPs and noninfectious subviral particles are generated. The subviral particles can compete with VLPs to interact with neutralizing antibodies. For studying viral antigen/antibody interactions, the favorite ratio of infectious VLPs to noninfectious subviral particles should yield results that satisfy the assumptions of law of mass action (1, 12, 21). To test weather the VLP-derived results follow the law of mass action, we performed the neutralization assay using various FFU of flavivirus VLPs and different dilutions of WNV-immune serum. No significant difference in neutralizing titer was observed with increasing dilutions of VLPs (data not shown). These results indicate that the VLP-based neutralization assay satisfies the law of mass action.

Figure 4. VLP-based neutralization assay. Vero cells (4×104) were seeded in each well of 96-well plates on a day before assay performance. The WNV-infected mouse serum was heat inactivated at 56℃ for 30 min. Equal volumes of 10-fold diluted sera and flavivirus VLPs (100 FFU) were incubated at 37℃ for 1 h. The neutralized VLPs were used to infect Vero cells. At 22 h p.i., the infected cells were subjected to luciferase assay. The percentages of neutralization were determined by the luciferase signals following the formular: Neutralization percentage = [1-(luciferase activity with WNV-infected mouse serum)/(luciferase activity without serum)] × 100%.
Construction and characterization of flavivirus Rluc2A-Rep
Production of infectious VLPs for a panel of flaviviruses
Stability of luciferase gene in WNV VLP
Virus-type specific diagnosis of WNV infection using flavivirus VLPs
-
The most accurate method for detecting neutralizing antibodies against flavivirus is PRNT. Although the use of this method has allowed for great insight into flavivirus biology and pathogenesis, it has several limitations. The PRNT assay is labor-intensive, and is difficult to perform in large scale. The quantitative power of the PRNT is subject to the number of wells examined and the number of plaques counted manually by the investigator.
Moreover, the PRNT approach involves the use of live infectious virus, which requires higher biosafety level containment (21). In this study, we describe a VLP-based serologic approach to differentiate WNV infection from other flavivirus infections. In comparison to PRNT, the VLP-based neutralization method has several advantages, including speed, sensitivity, quantitative power, and safety. Although the VLP-based neutralization assay significantly improved the diagnostic performance of ELISA, PRNT, and IFA assay, we were unable to completely eliminate crossreactivity, which is the intrinsic nature of flavivirus neutralization. Nevertheless, our results clearly showed that WNV-infected mouse serum inhibited WNV VLP more specifically and selectively than the DENV-1 and YFV VLPs (Fig. 4).
The yield of DENV-1 VLP is significantly lower than the yields of WNV and YFV VLPs. The reason for lower titer of DENV-1 VLP might be the slower rate of RNA replication, slower VLP release, or more rapid decay of released VLP. It was recently reported that incubation of transfected cells at lower temperature allows for production of DENV-1 VLP to a significantly higher titer (2). In our hands, lowering the cell culture temperature to 30℃ did increase the DENV-1 titer from 2×103 to 1×105 FFU/mL (data not shown). However, the absolute luciferase light units derived from the DENV-1 VLP were much lower than those derived from the WNV and YFV VLPs, even when infections were performed at the same FFU per cell. As discussed above, the lower luciferase signal was due to the lower replication nature of DENV-1. However, since the neutralization efficiency is determined by the decrease in percentage of luciferase signal from VLP of the same virus type (not from the VLPs of different virus types), the lower luciferase signals from the DNEV-1 VLP are still valid for quantification of neutralization efficiency.
In summary, the results presented in this study has demonstrated the proof-of-concept that VLPs can be used for virus-type specific serologic diagnosis of flavivirus infections. The VLP-based neutralization assay has kept the "gold standard"of the PRNT assay. Importantly, the new assay has significantly improved the throughput, shortened the assay time, and lowered the assay requirements for biosafety containment.

 DownLoad:
DownLoad: