











-
Bluetongue (BT) is an infectious, non-contagious, arthropod-borne viral disease of domestic and wild ruminants. Sheep are the primary host but other ruminants are also affected. The disease is reported from tropical, subtropical and temperate regions of the world and recently also from many European countries [24].Culicoides midges are the vectors for bluetongue virus (BTV). In India, 11 serotypes are reported to be isolated and the existence of another 10 types is based on sero-surveillance [22]. BT is one of the economically important diseases of sheep and is notifiable to the World Organisation for Animal Health [Office International des Épizooties (OIE)] due to its potential for rapid spread and socio-economic impact. Presence of BT in a country can restrict animal and animal by-product movement, thereby interfering with international trade. The aetiological agent, BTV is the prototype of the genus Orbivirus of the family Reoviridae [4]. BTV is a nonenveloped virus consisting of 7 structural (S) and 4 non-structural (NS) proteins and it has a double-stranded RNA (ds-RNA) genome of 10 segments.
Bluetongue is enzootic in India and it occurs sporadically. Details on its prevalence, economic impact, vector biology and the genetic information on Indian isolates are scanty. Moreover, presence of multiple serotypes in the country has complicated the diagnosis, serotyping, virus characterisation and the implementation of control strategies. A nation-wide network project on bluetongue (AINP-BT) was launched in 2000, with the goal of developing diagnostics and vaccines. In addition, the epidemiology, disease prevalence; vector biology, serotyping and genetic characterization of BTV isolates are being studied. Currently, the sero-surveillance and sero-diagnosis of BT is being carried out by detection of group-specific anti-BTV antibody in sera using imported agar gel immunodiffusion (AGID) and enzyme linked immunosorbent assay (ELISA) kits. This importation is not economical and increases the risk of importing exotic pathogens.
Under field conditions, BT diagnosis relies on the epizootiology, vector distribution, clinical symptoms and pathological lesions. The clinical symptoms and pathognomonic lesions are found in sheep and some other ruminants while other ruminants can be asymptomatic hosts. Tests to identify the viral antigens in the tissues/body fluids or antibodies in sera are available, such as AGID, serum neutralisation (SNT), fluorescent antibody (FAT), or ELISA. The AGID has been the most common test used for the detection of group-specific anti-BTV antibodies in sera as it is relatively simple and economical. However, it cross-reacts with other orbiviruses, particularly epizootic haemorrhagic disease virus of deer (EHDV) and the test is not quantitative. This has furthered the development of MAb based competitive/sandwich ELISAs for the detection of antibodies to BTV or BTV antigen [2, 9]. Recently, the use of the polymerase chain reaction (PCR) and quantitative PCR for detection of viral nucleic acid have facilitated BT diagnosis [14]. PCR may be used for sero-grouping of orbiviruses and, serotyping and topotyping of BTV isolates. The criteria for selection of assays for the diagnosis of BT include specificity, sensitivity, speed, cost and availability of expertise. The antigen-capture ELISA (Ag-ELISA) to detect BTV antigen directly from blood or clinical samples is an efficient tool for the large-scale screening and requires minimal technical expertise and equipment. It has been reported that the polyclonal antibody based antigen capture ELISA was insufficiently sensitive to detect BTV antigen directly in blood samples [5, 11]. However the MAb based sELISA is more sensitive and specific and can be used to identify viral antigen in blood or other clinical samples [23]. In this study, a sELISA was used to detect the BTV group-specific antigen.
HTML
-
BHK21 (ATCC, Manassas, Virginia) cell line was used for production of BTV. Cells were grown at 37℃in Eagle's minimum essential medium (EMEM, M/s Sigma, St Louis, Missouri) supplemented with 10% newborn calf serum (NBCS, M/s Hyclone, Logan, Utah), and antibiotics (benzyl penicillin and streptomycin sulfate @ 100 IU and 100 µg per mL, respectively). The cells were maintained in the maintenance medium containing 1% NBCS. The BTV-1, 2, 15, 17, 18 (Rahuri) and 23 (Dehradun) serotypes available in the repository, Division of Virology, Indian Veterinary Research Institute (IVRI), Mukteswar, Nainital(District), Uttarakhand were used in this study. These serotypes were amplified and maintained in BHK21 cell culture. Hybridoma clones were produced using BTV 23 whole virus as immunogen. In the present study, different hybridoma clones namely, 4A7, 4A10, 4A11, 4C8, 4G9, 4H11, 5B5 and 5D5 reacted against purified whole BTV23 and recombinant VP7 of BTV23 in indirect ELISA.
-
Healthy guinea-pigs (n=4) each weighing 250-500 g, rabbits (n=4) each weighing 1-1.5 kg and sheep aged 6-12 months of either sex were used for raising hyper immune serum (HIS) against BTV23. These sera were used in sELISA either for capturing or detecting antigen. Rabbits, guinea pigs and sheep were immunised with purified BTV 23 or rVP7, as previously described [12]. After confirming the presence of BTV23 specific antibodies by AGID, sera from each species was collected and preserved at -80℃.
-
Truncated recombinant VP7 (rVP7) protein of BTV23 expressed in E. coli was purified using a nickle (Ni) column, as described elsewhere [15] and used for producing HIS in rabbits as well as for initial screening of a panel of VP7 reactive hybridoma clones in indirect ELISA (iELISA).
-
Confluent monolayers of BHK21 were infected with 0.01 multiplicity of infection (m.o.i) of each of the BTV serotypes separately in 150 cm2 tissue culture flasks; the cultures were incubated at 37℃. After 72 h post infection (PI), when 80% CPE was observed, infected cells were harvested and stored at 4℃. Double stranded RNA from cell cultures infected with BTV was extracted and analyzed in PAGE to confirm the presence of BTV [19].
-
For bulk production of BTV, BHK21 cells were grown in roller culture bottles (1 700 cm2, M/s Greiner, Germany); when 90% confluent, they were infected with BTV by adsorption for 30 min at 37℃. The virus was harvested and titrated, after 72 h PI when 80% CPE was observed [18]. Harvested virus was purified and stored at 4℃ [12].
-
Initially, a panel of MAbs secreting BTV specific antibodies was selected using indirect ELISA [1]. In brief, the ELISA plates were coated with purified BTV-23 antigen (1:50 dilution in PBS, pH 7.4) @100 µL/well. The plates were incubated at 37℃ for 1 h under constant orbital shaking. Unbound antigen was washed off using 2.5mmol/L PBS containing 0.05% Tween 20. Then, 100 µL/well of each culture supernatant of individual clones diluted 1:2 in blocking buffer (PBS + 0.01% Tween 20 + 1% BSA) were added in duplicate wells. Then plates were incubated at 37℃ for 1 h under constant orbital shaking and then washed. Anti-mouse horseradish peroxidase (HRPO) conjugate diluted 1:1 000 in blocking buffer was added at 100 µL/well. Plates were further incubated for 1 h as described earlier. Plates were washed and ortho-phenylenediamine (OPD) comprising 30%H2O2 (0.5 µL/mL of OPD) were added to all the wells. When the colour reaction was observed for 15 min, the reaction was stopped with equal volume of 1 mol/L H2SO4. The optical densities were read at 492 nm. The clones that gave significantly high absorbance (A492) values (at least two times greater with purified BTV23 than that of BHK21 antigen) were selected for further investigation. Similarly, BTV reactive clones were checked for their reactivity with rVP7 protein of BTV.
-
After checking the MAbs in sELISA, suitable clones was selected from a panel of MAbs and the cells secreting a particular antibody were subjected to single cell cloning three times, as described in the touchstone procedure [16]. Then the clone was transferred to a cell culture flask and amplified. The supernatant was collected and clarified at 800 × g for 10 min and preserved at –20℃. The isotype of BTV rVP7 reactive MAbs viz., 4A7, 4A10, 4A11, 4C8, 4G9, 4H11, 5B5 and 5D5 were determined using hybridoma cell culture supernatant, employing a commercial isotyping ELISA kit as per the manufacturer's protocol (M/s Sigma-Aldrich, St. Louis, USA). The titration of MAbs was performed as described by Singh et al. [21] with the following modifications. In brief, the ELISA plates were coated with the purified BTV23 [1:50 dilution in PBS (pH 7.4) @100 µL/well or rVP7 at 100 ng/well in 100 µL PBS]. The plates were incubated at 37 ℃ for 1 h under constant orbital shaking.Unbound antigen was washed off using diluted PBS (1:4 in distilled water). For determination of antibody titre, twofold dilutions of hybridoma culture supernatants were prepared in blocking buffer (1% BSA + 0.05% Tween 20 in PBS, pH 7.4) in separate eppendorf tubes; 100 µL /well was added to each designated well. The remaining steps are as described earlier. The dilution of the MAb which gave 75% A492of the plateau (maximum A492) was considered as the titre of each MAb. The SDS–PAGE was performed using the discontinuous buffer system [8] to demonstrate the specificity of MAb with group-specific VP7 protein of BTV, followed by Western blot [20].
-
Initially, a panel of MAbs viz. 4A7, 4A10, 4A11, 4C8, 4G9, 4H11, 5B5 and 5D5 reacting with rVP7 were screened for their suitability using rVP7, MAb (5B5) as the capture antibody, purified BTV-23 as positive antigen and mock infected BHK21 cells as negative antigen. VP7 specific HIS raised in rabbits was used as the detection antibody. The optimum dilutions of these reagents were selected by chequerboard titration. Then, sandwich ELISA was performed according to the procedure described by Portanti et al. [17] with the following modifications. 100µL of monoclonal antibody cell culture supernatant diluted 1: 700 in calcium and magnesium free PBS (pH 7.4) (M/s Sigma Corp., St. Louis, USA) was used to coat each well of the ELISA plate and the plates were incubated for 1 h at 37℃ under continuous shaking or kept at 4℃ overnight. The unbound capture antibodies were removed by washing three times. 50µL of blocking buffer (0.5% gelatin + 0.5% BSA + 0.05% Tween 20 in PBS, pH 7.4) was added to all the wells except the antigen blank wells to which 100 µL of blocking buffer was added. Clinical samples dissolved in 50μL PBS was added to duplicate wells. 50µL of known positive and negative antigens were added to 4 wells. The plates were incubated for 1 hr under constant shaking at 37℃ and were then washed and gently tapped on filter paper to remove any unbound sample. Washing was repeated after every step until the conjugate step. Then 100 µL of detection antibody (HIS rVP7) was added, at 1:500 dilutions in blocking buffer, to each well and plate was again incubated for 1 h at 37℃, with continuous shaking, and then washed. 100µL of anti-rabbit antibodies conjugated to HRPO was added to each well (1:2 000 in blocking buffer) and the plate was incubated for 1 h at 37℃ under continuous shaking. 100 µL/well of OPD (0.5 µL of 30% H2O2/mL of OPD) was added to all the wells and the plate was incubated for 10-20 min or until colour developed in positive wells or started to developing in negative antigen wells. Then the reaction was stopped by adding 100 µL/well of 1 mol/L H2SO4 to all the wells; the plate was read at A492 in an ELISA reader. The positive controls (infected tissue culture supernatants) and negative controls (mock infected tissue culture supernatants) were included in the test. The samples were classified as positive when the OD492 nm was equal to or greater than twice the OD of the negative control (p: n≥2).
Specificity of antibody secreted by clone # 5B5 was ascertained by testing the MAb against different viruses, such as goatpox virus (GTPV, Uttarkashi), sheeppox virus (SPPV, Srinagar), Orf virus (ORFV, Mukteswar), PPRV (Sungri) and foot and mouth disease virus (types O, A., C and Asia 1). These viruses were available at the Division of Virology and Project Directorate on FMD Laboratory (PD-FMD), IVRI, Mukteswar, India. However, the specificity of the MAb was not checked against EHDV as the virus was not available. Finally, sensitivity of MAb-based sELISA was assessed using infected cell culture supernatant and was the minimum number of virus particles that would give a positive result. The cell culture grown BTVs were BTV-1, BTV-2, BTV-15, BTV-17, BTV-18 and BTV-23 and were the only serotypes available in India. This test was carried out as described above. The available BTV serotypes were serially diluted twofold (log 2).
-
Sheep (n=9) that were negative for antibody against BTV and had not been vaccinated nor infected with BTV were selected for experimental infection. The sheep were housed in an insect proof small animal shed for 1 week before starting the experiment. Then animals were grouped into three categories of three animals each. Animals in group Ⅰ received BTV-18 and animals in group Ⅱ received BTV-23. Inoculation was by the intravenous route; 1 mL of cell culture propagated virus was given to each animal. The animals in group Ⅲ were kept as healthy in-contact control. Samples were collected daily from all the animals until day 15 PI and then every other day. The samples were collected, as described elsewhere [17], in 10 mL vacuatainer tubes (Vacutainer, Becton Dickinson) with and without ethylenediamine tetraacetic acid (EDTA). In addition, samples were collected at 15 and 28 days for serological study. Whole blood treated with EDTA, were fractionated using Histopaque (as per manufacturer's protocol), then plasma and buffy coat fractions were collected and stored at 4℃ for further testing. Red blood cells (RBCs) were washed three times by centrifugation in PBS (pH 7.4) at 1 260 × g for 10 min and stored at 4℃ for antigen detection. Blood without EDTA was allowed to clot and then centrifuged at 1 260 ×g for 10 min; serum was separated and kept at –20℃ until further use. To evaluate the test, a total of 102 blood samples from animals suspected of having been exposed to BTV were collected from different outbreaks occurred at S K Nagar, Gujarat and Satara, Maharashtra (India). Some of the samples were randomly selected and were passaged in the BHK21 cell line for 2-3 blind passages. Then the samples were diluted 1:1 in blocking buffer and tested using the optimized sELISA.
Cells, viruses and hybridoma clones
Experimental animals and immunisation
Recombinant VP7 protein
Identity of bluetongue virus
Bulk production of BTV 23 and purification
Reactivity of MAbs with bluetongue virus and recombinant BTV-VP7 protein
Bulk production and characterization of MAb
Optimisation of sandwich ELISA
Evaluation of the assay using experimental clinical/field samples
-
In this study, a MAb-based sELISA has been developed for the detection of group specific BTV antigen from clinical samples. Hence, six BTV serotypes found in India, BTV-1, 2, 15, 17, 18 and 23, were bulk purified and were identified based on RNA-PAGE. Ten discrete ds-RNA bands (L1, L2, L3, M1, M2, M3, S7, S8, S9 and S10) were obtained for each serotype in RNA-PAGE [3] (data not shown).The titre of BTV-23 was 106.5TCID50/mL and purified virus content was 1.75 mg/mL with 90% purity.
The VP7 protein of BTV is group-specific and conserved amongst all 24 BTV serotypes. Detection of group-specific antigen in clinical samples or the antibody to it in serum is an indication of either recent infection or sero-conversion, respectively. Therefore, eight MAbs, viz., 4A7, 4A10, 4A11, 4C8, 4G9, 4H11, 5B5 and 5D5, have been verified for their suitability for the sELISA. These MAbs were selected based on their reactivity with BTV (Fig. 1A) and with the highly purified truncated rVP7 protein (Fig. 1B).Only eight clones were found to react with the rVP7 protein in the indirect ELISA.
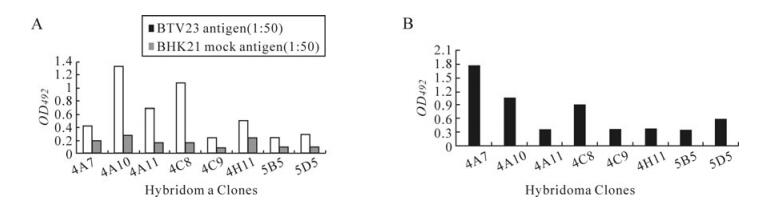
Figure 1. Reactivity of MAbs with whole virus(A), rVP7(B) in iELISA. A: Reactivity of hybridoma clones with BTV antigen and mock antiegn. B: Reactivity of hybridoma clones with rVP7 in indircet ELISA.
-
The selected clones were subjected to single cell cloning [16] and were amplified and preserved under liquid nitrogen. The MAbs obtained were further characterised using a commercial isotyping kit and the majority of the MAbs were IgG. However, three monoclonals, 4A7, 4G9 and 5D5, belonged to the IgM class. The selected MAbs were titered using purified BTV antigen and rVP7 protein in the indirect ELISA. Overall, the MAbs with sigmoid curves, 4A7, 5D5, 4A10, 4A11 and 4H11, had high antibody titres followed by the MAbs with linear curves, 4G9 and 5B5. The lowest antibody titres were observed in MAbs having concave reactivity curves either with BTV or rVP7 protein (4C8, 4A11, 4H11, 4A7, 4A10, 4G9 and 5B5) which reacted if undiluted and rarely when diluted 1:2. The details of the properties of each MAb are described in Table 1, Fig. 2. In order to ascertain the MAbs specificity with VP7 protein, all the MAbs were checked by blotting and were reacted with whole virus as well as rVP7, which yielded 38.24 kDa proteins. As our objective was to optimize sELISA, only the 5B5 data is shown here.

Table 1. Characteristics of bluetongue virus-specific monoclonal antibodies
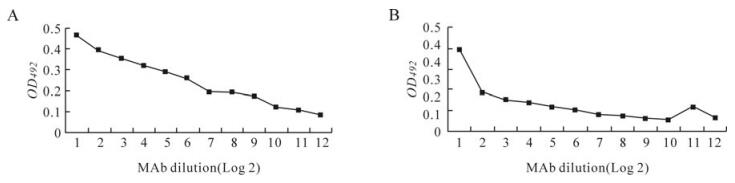
Figure 2. Titre of clone 5B5# with BTV (A) and rVP7 (B).
Taking advantage of the reactivity of these clones with rVP7, titre, isotype and Western blotting, the MAbs were checked for their suitability for sELISA in the detection of group-specific BTV antigen (VP7) in a chequerboard titration (data not shown). Only the 5B5 MAb was found suitable for the sELISA. The optimum dilutions of capture antibody (MAb, 5B5), detection antibody (rabbit HIS against rVP7) and purified BTV antigen were 1:700, 1:500 and 1:100, respectively. Mock infected BHK21 cell lysate was taken as a negative control and cut off was calculated as double the OD of the negative plus a standard deviation and was found to be 0.190. The test samples, which were greater than the cut off, were considered as positive. This MAb was directed against BTV VP7 protein and reacted with six serotypes of BTV (1, 2, 15, 17, 18, and 23) and rVP7 protein with varying intensities, and could also be used in Western blotting. The other BTV serotypes could not be tested as they are not available in India. The MAb belonged to IgG2a isotype and therefore, 5B5 was explored further. Some of such characteristics of MAbs have been studied by other investigators for optimising ELISAs [17]. In this study, the MAb was the capture antibody and rabbit HIS against rVP7 was the detection antibody and is in agreement with the previous reports [6, 23]. After optimizing the dilutions of the reagents, viz. capture antibody (MAb), detector antibody, positive and negative antigen, the test was further validated using experimental sheep (infected with BTV-18 and 23) and field samples.
Further, the specificity of the MAb # 5B5 was checked with other viruses in sELISA but no cross-reactivity was observed with these viruses (Fig. 3). All BTV serotypes were detected in undiluted infected cell culture supernatant and in the diluted supernatant. The BTV sELISA was considered positive when the positive /negative ratio was≥2. In the present study, detection limit of the assay varied with the serotype of BTV; this is in agreement with the earlier reports [23] (Table 2). Though the preliminary results are encouraging, the purification of the MAb and sensitivity of the test need to be verified with other BTV serotypes which are not available in India. Similarly, sELISA was successfully employed to detect the five BTV serotypes after amplification either in cell culture or embryonated chicken eggs (ECE) [11, 17]. This was similar to a report by Stanislawek et al. [23] that 17 serotypes of BTV could be detected directly from clinical samples. Hawkes et al. [5] was successful in detecting 24 serotypes of BTV after amplification in ECE. In this study, the sensitivity of the assay for six serotypes was studied. The specificity of the assay was not determined for EHDV as this disease has not been reported from India and virus is not available.

Figure 3. Specificity of sELISA. A1-H1: Antigen blank; A2-D2: Reference +VE; E2-H2: Reference –VE; A3 & B3: GTPV; C3 & D3: SPPV; E3 & F3: ORFV; G3 & H3: PPRV; A4 & B4: FMDV (O); C4 & D4: FMDV (A); E4 & F4: FMDV(C) and G4 & H4: ASIA-1.

Table 2. Results of bluetongue virus infected cell cultures tested with the MAb based sELISA
-
The assay was evaluated using experimental samples collected from sheep infected with BTV-18 and BTV-23. Infected animals had rise in temperature (Fig. 4), reddening, swelling, hemorrhages and epithelial erosion of face, lips, gums and tongues for 10 days. An overall positivity of 43.7% and 29.2% was observed, respectively, against BTV18 and BTV23 (Table 3).
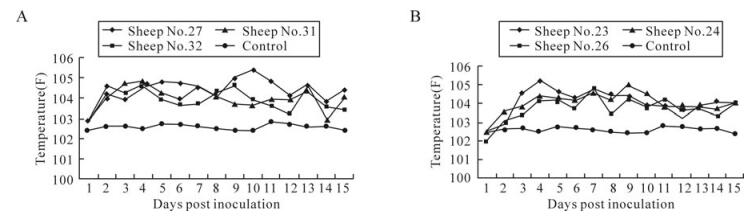
Figure 4. Experimental infection of sheep withBTV18 (A) and BTV23(B).

Table 3. Sandwich ELISA results of samples collected from experimental animal infected with BTV-18 and BTV-23
Also, the MAb performed well with clinical samples of washed RBCS, blood, buffycoat and plasma. The test could detect BTV antigen as early as, day 8 from blood, but rarely on day 7; antigen was detected until day 12 PI, this period corresponds to peak viraemia [7]. The control animals did not secrete any detectable amount of antigen either in blood or secretions. The group specific antigen could not be detected between days 0 to 6 and after day 12 PI. A total of 102 clinical samples (18 blood and 84 cell culture amplified) were tested using the sELISA. Of them, 29.4% were positive. The cell culture amplification improved the sensitivity of the test and as a result more positives were obtained.
Autopsy specimens or specimens from aborted foetuses like liver, bone marrow, brain, spleen, lymph nodes and lungs are suitable for virus isolation/ detection. Similarly, biopsy samples from the mucous membrane of digestive and respiratory tracts, vascular, skeletal and reticuloendothelial systems, blood, semen, oral and labial mucosa [10] are also suitable for isolation/detection of BTV. Therefore, in this study clinical samples like blood, washed RBCs, buffy coat, and plasma, collected from sheep experimentally infected with BTV-18 and BTV-23 were tested using the sELISA. The samples from days 0 to 15 PI were tested and the peak viraemia was observed on days 6 to 9 PI. The test detected antigen in all types of clinical samples from days 7 to 10 PI, as reported earlier [7, 23]. After sero-conversion, the sELISA failed to detect VP7 protein as the VP7-specific antibody blocked the binding of MAb to the BTV sero-group reactive protein. There was a decline in detectable BTV antigen and the presence of BTV as neutralizing antibody increased. All the inoculated sheep were sELISA positive, as shown in Fig. 3; this confirms the sheep were infected with BTV-18 and BTV-23 serotypes. The sELISA was optimised using the capture MAb, 5B5, which had a high titer that was specific to rVP7; this confirms the findings of Portanti et al. [17]. The MAb performed better as a capture antibody than the antibody produced in rabbits. The titer of BTV was higher in the RBCs than in whole blood or other clinical samples. The BTV has been reported to be more closely associated with RBCs than with other blood components and consequently protected from antibody and other defensive mechanisms. The virus is released upon the natural degradation of RBCs [13]. The life span of RBCs in sheep is less than cattle; consequently, the virus persists longer in cattle. The performance of the assay was improved by lysing the RBCs, either by sonication, or by treating with distilled water or 0.8% ammonium chloride.
In conclusion, the optimised sELISA is specific, rapid and suitable for the diagnosis of acute BT. The test can be used in all age groups of sheep and cattle in a non-enzootic area or to confirm an acute infection. However, the post infection group-reactive antibodies could limit the use of the sELISA in the diagnosis of BTV infection. The diagnostic efficacy of sELISA can be enhanced with due consideration of clinical signs, serology and PCR. Further, the optimized assay needs large scale sample evaluation.

 DownLoad:
DownLoad: