











-
Potato virus Y (PVY) is a member of the genus Potyvirus [15], the largest group of plant viruses. PVY occurs as long, flexuous particles measuring 730×11 nm, containing single-stranded, positive polarity RNA [6] with a genome size of approximately 9.7 kb. Potato and many other solanaceous crops like tomato and tobacco are infected by this virus. Potato is considered as world's fourth most important crop and infection of PVY on potato results in severely depressed yields. In Pakistan, the losses caused by PVY are estimated to be 40%-70% [19]. Depending on cultivar and viral strain, the symptoms induced by PVY vary from an almost imperceptible mosaic pattern up to severe necrosis and premature death of plants [25]. Control of PVY infection is difficult in potato because it is propagated vegetatively, which makes primary viral infection more destructive and persistent generation after generation.
Currently none of the available high yielding com-mercial varieties or advance potato lines in Pakistan has shown durable resistance against viruses [21]. Several strategies have been tested so far which include genetic modifications of the PVY coat protein (CP) gene or P1 gene [9, 16, 24]. As a result, in virus resistant plants, resistance is protein mediated and is was neither effective nor stable as the resistance conferred by gene silencing [5]. RNAi/PTGS in transgenic plants is an epigenetic form of RNA degradation and is associated with defense against viral infection and regulation of expression [13]. Evidence suggests that RNA viruses can generate double-stranded RNAs similar in sequence to the transcribed region of target genes, which then undergo endonucleolytic cleavage to generate small interfering RNAs (siRNA) that promote degradation of cognate RNAs. Small interfering RNAs (siRNAs) are of 21 nt and have been reported to play a crucial role in RNA silencing.
Although the siRNA gene-silencing approaches show great promise towards virus-resistance in plants, some limitations still exist e.g. at present there is no well-defined set of rules for designing siRNA oligonucleotides. Because some candidate siRNAs may work well and others may not, a panel of suitable siRNAs must be tested empirically to find potent one. To take full advantage of gene silencing by siRNA, an appropriate screen should be devised. The screening can be done by cloning of each siRNA and analyzing expression knockdown of the target gene in cell culture system, which is very laborious and time consuming [32] or by screening based on phenotype conferred by knockdown of the target gene. To overcome these laborious methodologies, we have optimized an efficient screening technique for siRNAs using transient expression in mammalian cell lines.
HTML
-
For the potential and effective knockdown of PVY in-vitro, a fragment of CP-PVYgene conserved among all four PVY strains (PVYNTN, PVYN, PVYo and PVYC) was used for primer design. For this purpose, multiple sequence alignment was performed using full length CP gene sequence of all four strains; PVY NTN (Accession # GU550076), PVY N (Accession # AJ890342), PVY o (Accession # AJ890349) and PVY C strain (Accession # AJ890348). The conserved sequence was then used to design primers PV7 and PV8 (Table 1) for the amplification of a 480bp CP gene fragment. The PCR product was cloned initially into pCR2.1-TOPO (Invitrogen, USA) for sequencing and then re-amplified by introducing Hind Ⅲ and BamH Ⅰ sites in both reverse and forward primers (PV9 and PV10, Table 1). Amplified fragment was gel-purified and inserted into the mammalian expression vector, pCDNA 3.1(+) (Invitrogen, USA), to obtain the plasmid, pCPVY, the cDNA clone of CP-PVY. E. coli DH5α competent cells were transformed and orientation of insertion of cloned fragment was confirmed by restriction analysis.

Table 1. Sequences of primers used in amplification, cloning and real-time PCR studies of CP-PVY mRNA
-
Small interfering RNAs (siRNAs) against CP-PVY RNA were designed by selecting a 50-100 nucleotide downstream region of the start codon with 35%-50 % G+C content. Stretches of 4 or more nucleotide repeats were avoided, and sequences that share homology with other related or unrelated genes of the same organism were also avoided. To design siRNAs, software provided by Ambion, (USA) was used. The designed template oligonucleotides (DNA) for each siRNAs were custom synthesized from Sigma Aldrich (Germany). All oligos were 29 bp in length with 8 nucleotides of the T7 promoter sequences added at the 5' end for final synthesis of duplex siRNAs (Table 2). Sense and antisense oligo templates were annealed to synthesize siRNAs using a siRNA construction kit (Ambion, US) according to the user manual.

Table 2. Sequences of siRNAs designed against capsid protein gene of PVY used in the study
-
The CHO-k cell line was kindly obtained from Biopharmaceuticals lab (CEMB, Lahore Pakistan) and were grown in DMEM media with 100μg/mL of strep-tomycin, 100U/mL of penicillin and 10 % FBS (Gibco).
-
CHO-k cells were seeded in 6-well plates at 1×106cells/well and were grown overnight prior to transfection. Cells were transfected at 50%-70% confluence containing 1.5 mL of medium per well. Cells were co-transfected with pCPVY construct, together with siRNAs. All transfections were performed using lipofectamine 2000 as transfection reagent (Invitrogen, CA). For knockdown of CP-PVY mRNA, 50ng to 1μg pCPVY was used while the siRNA concentration was kept at 100nmol/L. All transfection experiments were done in triplicate.
-
RT-PCR was used to measure the mRNA expression during the knockdown study of CP-PVY. Total RNA was isolated from CHO-k cells by using TRIzol reagent (Invitrogen). cDNA was synthesized using 1μg of total cellular RNA treated with DNase, oligo dT primer and dNTPs. After being denatured at 70℃ for 5 min and then cooled to 37℃, 40 U of M-MuLV (Fermentas) were added and the extension was carried out at 37℃ for 1 h. Reaction was stopped by heating at 70℃ for 10 min. PCR was carried out with the corresponding PV9 and PV10 primers (Table 1). The PCR amplification profile was adjusted at 27 cycles: denaturation at 94℃ for 45 s, annealing at 58℃ for 45 s, and extension at 72℃ for 45 s. All samples were run in triplicate and normalized to GAPDH mRNA.
-
For the real-time PCR, Primer3 software provided by (http://frodo.wi.mit.edu/primer3/verifiedon2009/07/22) was used to design specific RT-F and RT-R primers (Table 1), that could amplify a fragment size of 100bp out of the 480bp conserved CP-PVY gene portion. Tm was optimized for the primers and checked for absence of non-specific bands after electrophoresis in 2% agarose gel. Real-time PCR was performed on a Cepheid Smart Cycler Ⅱ using SYBR Green Mix (Fermentas). 1μg of cDNA was used in each reaction to study the knockdown efficiency of CP-PVY mRNA caused by siRNAs. The amplification profile was adjusted at 30 cycles: denaturation at 94℃ for 30 s, annealing at 58℃ for 30 s, and extension at 72℃ for 30 s after initial denaturation at 94℃ for 10 min. The GAPDH was used as a control for normalization. The relative gene expression analysis was done by using Ct values in different samples and calculated standard deviation. Each real-time PCR assay was performed in triplicate.
Construction of expression plasmid
Design and synthesis of siRNA
Cell culture
Transfection
Reverse transcription PCR analysis
Real-time PCR analysis
-
Multiple sequence alignment of all four reported PVY strains namely PVYNTN (Accession # GU550076), PVYN (Accession # AJ890342), PVYo (Accession # AJ890349) and PVYC (Accession # AJ890348) was done using Clustal W (1.83) software. Primers were designed from the conserved portion in a way that they can amplify the CP gene fragment of 480bp from every strain of PVY. In the present study, we have amplified CP-PVY fragment of 480bp from local PVY isolate that shows 99% homology when compared with NCBI reported PVY sequences. Fig. 1 shows sharp amplification of 480 bp from local PVY isolate using primers PV7 and PV8 (Table 1).
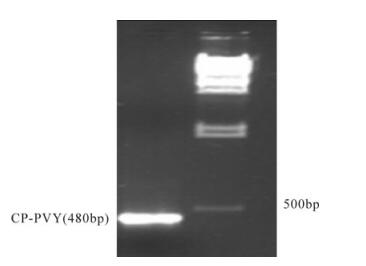
Figure 1. CP-PVY amplification from PVY local isolate. A 480bp gene fragment was amplified from local PVY isolate using primer pair (PV7 and PV8) which was designed from CP gene conserved among all four PVY strains.
The 480 bp sequenced fragment of the CP gene from a local PVY isolate was used as template to design siRNAs. In total, six siRNAs were designed against the CP-PVY region starting from 150nt-to 630nt-out of the complete 800bp CP gene. ssDNA oligos were transcribed into ssRNA oligos separately which, upon mixing, gave dsRNA oligos ready to use. Fig. 2 shows the dsRNA oligos (siRNAs) electrophoresed onto 20 % PAGE (polyacrylamide gel electrophoresis) showing a compact band which indicates the synthesized oligos were intact and had not been degraded.
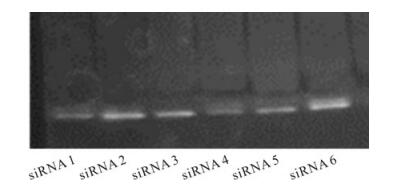
Figure 2. Polyacrylamide gel electrophoresis of synthesized duplex siRNAs. 20% polyacrylamide gel was prepared for resolution of siRNAs (dsRNAoligos) targeted against CP-PVYmRNA to check their integrity and estimate concentration.
-
In the CHO-k cell line, we transiently expressed CP-PVY mRNA from a cloned plasmid, pCPVY. Mammalian expression construct, pCPVY, containing a cDNA clone of the CP-PVY fragment was transfected in CHO-k cells at various concentrations ranging from 50 ng to 1 μg. Assay time was kept at 24 h. Results demonstrated that CP-PVY mRNA was expressed constitutively in transfected cells with expression starting from 50ng of pCPVY (Fig. 3). It was also observed that RT-PCR expression increases proportionally with increase in pCPVY concentration as seen in fig 3. Neither CP-PVY mRNA expression was observed in cells transfected with 1μg of empty vector pCDNA3.1(+) nor in non-transfected cells.
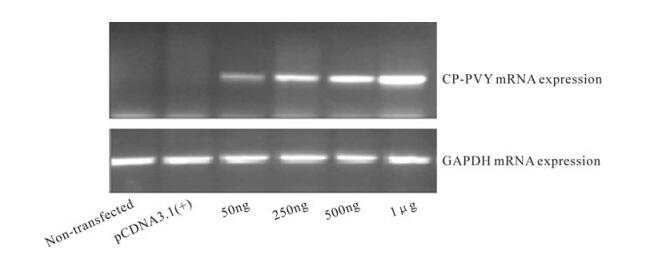
Figure 3. CP-PVY is expressed and active in CHO-k cell line. The indicated cell line was used for RNA extraction after 24 h transfection with expression vector pCPVY at different DNA concentrations ranging from 50 ng -1 µg. GAPDH was used as internal control. Total cellular RNA was extracted after 24 h post transfection and subjected to reverse transcriptase PCR using gene specific primers. In the figure CP-PVY mRNA depicts onstitutive expression in CHO-k cells.
For transfection, lipofectamine (transfection reagent) concentration was optimized. Best results were obtained at 10μL of lipofectamine per 6-well culture plate for 50ng pCPVY. So based on optimization results for DNA and transfection reagent, 0.05μg pCPVY DNA along with 10μL lipofectamine per 6-well was found to be the best combination to obtain optimum expression in CHO-k cells.
-
During knockdown studies of CP-PVY mRNA, 100nmol/L concentration of siRNA was combined with optimized transfection conditions. Data obtained showed that all six siRNAs used reduced the mRNA expression of target gene to some extent but only siRNA1 significantly reduced CP-PVY mRNA expression by up to 12.25 fold and, as is clearly shown in Fig. 4A, expression was almost diminished or very faint in cells transfected with siRNA1 as compared to the control which consisted of scrambled siRNA along with with 50ng of pCPVY. The remaining siRNA knockdown values were: siRNA 2 -7× decrease; siRNA 3 -8× decrease; siRNA 4 -10.8× decrease; siRNA 5 -9× decrease and siRNA 6-10× decrease (Fig. 4B). These values were based on Ct values obtained from real-time PCR studies. The total RNA extracts derived from the siRNA transfected cells were also tested for the expression of GAPDH mRNA which was found to be similar in all CHO-k cells, non-transfected and transfected with pCPVY plasmid, verifying the results obtained. GAPDH transcript levels showed no change in non-transfected or transfected cells. Also, Real-time PCR data verifies conventional RT-PCR data, with almost same folds decrease in CP-PVY mRNA expression.
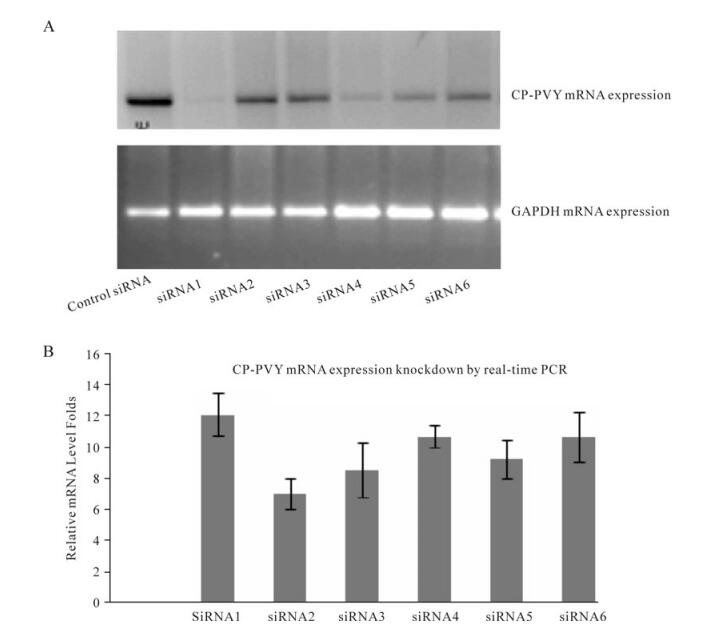
Figure 4. Knockdown of CP-PVY by siRNAs in Chinese hamster ovary cells. The CHO-k cell line cultured in 6-well plates were transfected with 0.05 µg of expression vector, pCPVY, and 100nmol/L of siRNAs. A: Inhibitory effect of capsid protein gene mRNA expression by siRNAs in CHO-k cell line analyzed by RT-PCR: shows the level of expression of CP-PVY mRNA in the cells transfected with 100nmol/L siRNAs 1-to 6-and 50ng of pCPVY. Total RNA extracts were subsequently used for GAPDH mRNA expression level as internal control. B: Inhibitory effect of CP-PVY mRNA expression by siRNAs in CHO-k cell line by real-time PCR: shows the knockdown level of CP-PVY mRNA expression mediated by siRNAs 1-6 in transfected CHO-k cells after 24 h transfection. GAPDH was used to normalize the samples. All measurements were made in triplicate in three independent experiments.
Multiple Sequence Alignment of CP-PVY gene
CP-PVY mRNA is constitutively expressed in CHO-k cell line
Capsid Protein RNAi specifically reduces CP-PVY mRNA expression in CHO-k cell line
-
The primary aim of the present work was uses siRNAs to efficiently engineer strong PVY-resistance in potato, an economically very important crop that is severely affected by this virus. To achieve this goal, we have developed a methodology to screen potential siRNA from a bulk panel because some candidate siRNAs may work well and others may not. Our methodology is based on transient expression in a mammalian cell line.
The capsid protein gene of PVY (CP-PVY) was selected to confer resistance against PVY as the CP gene is reported to interfere with PVY uncoating and translation and for transmission of potyviruses by aphids [1]. Moreover, several studies have shown that a fragment of the CP transgene confers resistance/ immunity in transgenic plants [5, 9]. Also, virus-derived dsRNA from transgenes can fully suppress viral infection through RNA silencing [14, 23, 29]. We selected a 480bp fragment of the CP-PVY gene as template to design six siRNAs. A segment of the CP gene was preferred over a full CP gene template because the multiple alignment of four NCBI reported PVY strain sequences showed a highly conserved region among the CP genes. In our study, we used this region to design primers for CP gene fragment amplification from a local PVY isolate that has 99% sequence homology with reported sequences in Genbank. Thus resistance conferred by the particular screened siRNA will be equally effective against all four PVY strains (PVYN, PVYNTN, PVYo and PVYC).
siRNAs were screened during transient expression assays of 24 h in mammalian cell line (CHO-k) and the siRNA (siRNA1) was found to knockdown CP-PVY mRNA expression by up to 12.25 folds when analyzed through real-time PCR. In our study, we used the CHO-k cell line instead of plant cells because this was a robust line, gives expression in 24 h compared to plant cells which need cloning of each siRNA out of a panel and transformation in plant followed by expression analysis which is time consuming and laborious. Also, our approach of using siRNA is based on concept of "RNA silencing of target genes" whose mechanism is conserved among all kingdoms including mammalian cells and plants alike and the unifying nature of this mechanism is the presence of 21-to 26-nucleotide (nt) small interfering RNAs (siRNAs) as supported by [10, 11, 20, 30]. Therefore, we used the CHO-k cell line for siRNA screening studies instead of plant cells.
RNA silencing refers to gene silencing involving a sequence-specific degradation of RNA. It includes post transcriptional gene silencing (PTGS), co-sup-pression, and RNA-mediated virus resistance in plants; RNA interference (RNAi) in animals; and silencing in fungi (quelling in Neurospora) and algae [20]. In addition, biochemical and genetic analyses have shown that the core mechanisms of RNA silencing are shared among different eukaryotes [2, 12, 17, 20, 28, 31].
Furthermore, knockdown by siRNA was specific to pCPVY, i.e. against the CP-PVY transgene while no RT-PCR/real-time PCR expression was observed in cells transfected with empty vector pCDNA 3.1 (+) or in non transfected CHO-k cells. The reason why resistance was effective against CP transgene only can be explained according to the concept of the homology-dependent gene silencing (HDR) mechanism [8, 22]. Our findings are consistent with already proposed mechanism of RNA silencing [4, 7, 18].
In summary, we have described the development of a methodology to screen one single potent siRNA from a large number of siRNAs designed where expression of targeted genes can be reduced substantially to influence biological functions. This technology builds on the use of mammalian cell culture system for transient expression combined with the use of RNAi to silence gene expression. The present methodology allows us to generate i) Culture conditions in which expression of specific gene(s) can be obtained constitutively, ⅱ) Screening for potent siRNA from bulk and ⅲ) Robust and precise results. We believe that the use of siRNA for stable RNAi in plants will become a powerful aid to probe gene function in-vivo and for gene therapy of diseases caused by viruses.

 DownLoad:
DownLoad: