











-
The virus-host interactions that is necessary for the infecting virus to undergo proliferation [9] are particularly evident in the Herpes simplex virus (HSV-1)[6, 12, 18]. As a double-stranded DNA virus, HSV-1 has a complicated gene structure and multiple structural protein components [13], making its in-teraction with host protein molecules markedly more complex. The interaction of HSV-1 with protein molecules of the host cells during infection is of great significance in affecting and regulating viral multiplication[4, 17].
Our preliminary work indicates that host cells show obvious changes in expression profiles in response to HSV-1 infection [7]. Among them, a number of new host protein molecules were up-regulated and exerted different transcriptional and regulatory functions in the course of HSV-1 infection and proliferation, [7, 19]directly affecting viral multiplication efficiency [7, 19]. Meanwhile, further analysis showed that after infection, some proteins participated in the structural assembly of the HSV-1 viral capsid [11]. Experiments in this study show that the host cell protein C9orf69, a protein of unknown function, promotes viral proliferation efficiency in the course of its interaction with the HSV-1 structural protein UL25, a viral capsid protein involved in viral DNA encapsidation and uncoating. This result, together with other preliminary work, suggests that the interaction of HSV-1 with the host cell involves the interaction of a variety of different proteins and macromolecules to regulate HSV-1 proliferation.
HTML
-
Cells and Virus
African monkey kidney cells (Vero) and human embryonic kidney cells (HEK293) were cultured in nutrient solution DMEM containing 10% calf serum at 37℃. HSV-1 virus (F strain), preserved by our laboratory, was proliferated in a monolayer of Vero cells.
Plasmid construction
Plasmids were constructed using standard methods. The UL25 encoding gene was inserted into eukaryotic expression shuttle vector pGBKT7 to construct the pGBKT7-UL25 plasmid for yeast two-hybrid. The C9 and UL25 encoding genes were respectively inserted into eukaryotic expression vector pEGFPC1 and pDsRedC1 to construct fluorescent pEGFP-C9 and pDsRed-UL25 plasmids, and inserted into prokaryotic expression vector pGEX-5X-1 to construct pGEX-C9 and pGEX-UL25 plasmids. The C9 encoding gene was inserted into eukaryotic expression vector pcDNA3 to construct pcDNA-HA-C9 plasmid and into pGE vector to construct pGE-C9 which expresses siRNAs directed against coding sequences of the C9orf69 mRNA (Table 1). All recombinant plasmids were detected and confirmed by enzyme digestion and sequencing. The plasmids of pGBKT7-Lam, pGBKT7-p53, pGADT7-T and pGE-Neg were stored in our laboratory.

Table 1. PCR primers used for the constructs
Library screening by yeast two-hybrid
pGADT7-T and the recombinant pGBKT7-UL25 were co-transfected into yeast strain AH109 using standard PEG/LiAc methods and equally coated onto DDO (SD/-Trp/-Leu) and QDO (SD/-His/-Trp/-Ade/ -Leu) flats and cultured at 30℃ for 3 to 5 days. Cytotoxicity and self-activation activity were determined through observing yeast clone growth and size.
Plasmid pGBKT7-UL25 was transfected into strain AH109 and fused with pre-prepared Y187 yeast (strain?) containing a human renal cell cDNA library according to instructions by Clontech [2]. Yeast strains were inoculated in 45 mL 2×YPDA/Kan liquid medium and cultured at 30℃, 50 r/min for 20 h. The thallus was washed with 50 mL 0.5×YPDA/Kan twice after centrifugation, re-suspended in 10 mL 0.5× YPDA/Kan liquid medium, coated on TDO (SD/-His-Leu-Trp) plates with 200 μL per plate, and cultured at 30℃. When clones matured, the single clones were transferred to QDO plates and cultured at 30℃ for 3 to 8 days.
β-galactosidase activity analysis
A single fused yeast clone was selected for inoculation in 5 mL DDO (SD/-Leu-Trp) at 30℃ and 200 r/min overnight, then transferred into YPDA medium for 3-5 h. Thallus was collected when A600= 0.5-0.8. The supernatant (after treatment with lysate) with a concentration of 4 mg/mL ONPG (Promega) was incubated at 30℃ until color appeared and the reaction was terminated with 1 MOL/L Na2CO3. β-gal activity units = 1000 × A420 / t × 5 × A600 were calculated and experiments were repeated three times to take the mean. Positive and negative controls were set at the same time.
Fluorescence position analysis
When Vero cells in six-well plates grew to 70% confluency, the eukaryotic expression plasmids (2μg/hole) encoding fluorescent proteins were transfected with Lipofectamine 2000 (Invitrogen). Slides were picked up 30 h after transfection and observed under a fluorescence microscope (Nikon inc). In addition, slides transfected with HSV-1 (MOI=1) 24 h later were picked up at different times and observed under a fluorescence microscope.
MTT assay
HEK293 cells transfected with plasmids pcDNA3 or pcDNA-HA-C9 were digested by trypsin 6 h after transfection to prepare a cell suspension which was inoculated into 96-well plates at 104 cells/well and cultured for 3 consecutive days at 37℃, 5% CO2 with saturated humidity. Six wells of each infected cells were collected to conduct tetrazolium (MTT) colorimetric detection [8].
Extraction of viral DNA or total RNA and realtime quantitative PCR (real-time PCR) analysis
HSV-1 virus DNA was extracted using a MiniBEST Viral RNA/DNA Extraction Kit (TaKaRa), and hydrolyzed with RNase (TaKaRa). HSV-1 standard samples with a concentration of 105, 104, 103, 102, 101, or 100 copy/μL were added to the same PCR plate to determine the standard curve. Primers used were as follows: UL35F: 5'-ATGGCCGTCCCGCAATT-3' and UL35R: 5'-CGAAGGTTCTCGAACGAC-3'.
Total RNA was extracted using Trizol-A+ (TIANGEN) and hydrolyzed with DNase (TaKaRa). Total cDNA was obtained by reverse transcription using Oligo-dT as primers for real-time PCR. β-actin was used as internal reference for relative quantitati-fication[21].
Real-time PCR was conducted in an ABI 7500; the protocol was conducted as described by the Prime-ScriptTM Real-time PCR kit (TaKaRa).
Antibody preparation and immunoprecipitation
BL21 strains for the transformation of plasmid pGEX-C9 and pGEX-UL25 were induced with 1 mmol/L IPTG to express the fusion protein GST-C9 and GST-UL25. Proteins were separated using SDS-PAGE electrophoresis, and proteins of corresponding molecular weight were excised and purified. Purified proteins were obtained by electric elution. The purified fusion proteins were mixed with an equal volume of complete Freund adjuvant emulsification. This was then injected subcutaneously into Kunming mice at multiple points in the abdomen and re-immunized at 2 and 4 weeks (with Freund incomplete adjuvant emulsification). Blood samples were collected from the eyeball 2 weeks after the third immunization to prepare C9orf69 and UL25 antiserum.
HEK293 cells were cultured in 10 cm cell culture plates and transfected with plasmid pcDNA-HA-C9, and 20 h later the cells were infected with HSV-1 (MOI = 20). Cells were harvested 16 h after infection, centrifuged 600×g at 4℃ for 10 min and the supernatant was discarded. Cells were lysed with RIPA lysate (150 mmol/L NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mmol/L Tris·HCl, pH 7.5), centrifuged at 14, 000×g, 4 ℃ for 5 min and cell debris were removed. A portion of the cell lysates were preserved for Western blot while the remaining lysates were added to anti-C9 (or anti-UL25) antibody by standard immunoprecipitation methods and incubated at 37℃ for 2 h. The immune complex was added to protein A agarose (Beyotime Institute of Biotechnology) at 4℃ for 1 h. The precipitated samples were used for SDS-PAGE gel for electrophoresis, and then transferred to PVDF membranes by standard methods. The anti-UL25 (or anti-C9) antibody was analyzed by standard Western blot[21]. Membranes were blocked with 5% BSA-TTBS after SDS-PAGE electrophoresis and protein transfer. Anti-UL25 or anti-C9 antiserum (1:100 dilution) was used as the primary antibody and IgG-HRP as the secondary antibody (1:5 000 dilution) for detection.
Flow cytometry
One hundred microliters of HEK293 cells were cultured at 37℃, and the cells were synchronized using the thymidine double block method after transfection with plasmids pEGFP and pEGFP-C9 respectively. Cells were collected at different times, washed once with PBS (pH 7.4), slowly added to 0.75 mL of cold ethanol and 0.25 mL PBS (pH 7.4) and fixed overnight at 4℃. The fixative was discarded, and 100 μL of 0.1% Triton X-100 was added for 10 min followed by addition of 100 μL of 0.1 mg/mL RNase A at 37℃ for 30 min. One hundred microliters of 0.25 mg/mL propidium iodide (PI) was added and removed from light at 4 C for 30 min. Cell cycle stage was determined by flow cytometry (BD).
Statistical analysis
Data were expressed as the means and standard deviations. Statistical significance between the treatment groups was calculated using the two-way ANOVA (GraphPad Prism), and a P-value of < 0.05 was considered significant.
-
Identification of host cell protein interactions with HSV-1 structural protein (UL25)
Based on the yeast two-hybrid system using the HSV-1 structural protein UL25 as the bait target molecule, a cDNA library from human kidney was screened to obtain a number of host molecules that may interact with protein UL25 (Table 2). Among them, C9orf69 was strongly detected. Though its function is unknown, informatics analysis shows that the protein is encoded in chromosome 9. Its mRNA length is 2866 bp, and the encoding frame is on nucleotides 452-859, consisting of 136 amino acids (Fig. 1). The molecule contains α-helical structures but lacks signal sequences, suggesting that it is not secreted.

Table 2. UL25 predicted proteins interactions by yeast two-hybrid in host cells
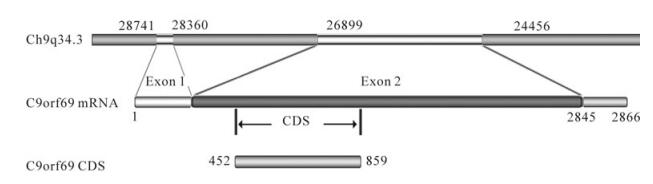
Figure 1. C9orf69 gene position and sequence characteristics
Identification of the interaction between C9orf69 and HSV-1 UL25
Based on the data that the C9orf69 protein may interact with UL25, we first conducted a β-gal activity assay that confirmed the interaction between C9orf69 and UL25 (Fig. 2A). The fused expression gene pDsRed-UL25 was cotransfected with pEGFP-C9, and confocal microscopy was used to confirm colocalization of the protein in the cell (Fig. 2B). The result confirmed the Y2H result that C9orf69 and UL25 interact in the cell. We also used cell cultures infected with HSV-1 and transfected with pcDNA-HA-C9 for immunoprecipitation which further confirmed the interaction between C9orf69 and UL25 exists in the course of a HSV-1 infection (Fig. 2C).
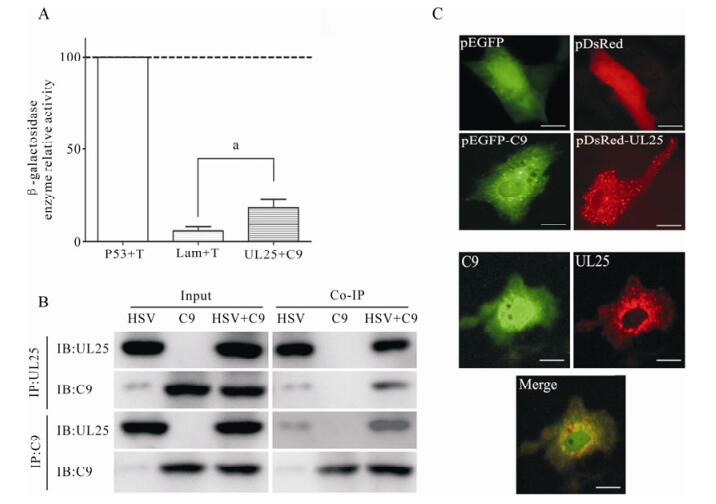
Figure 2. Determination of the interaction between C9orf69 and UL25. A: Detection of β-gal activity. Yeast colony for plasmid transfection were picked and cultured in minimal culture medium; A600 was recorded after harvest. Supernatant after cell lysis was treated with ONPG, terminating the reaction as the yellow color appears. Reaction time was recorded, and A420 was measured by β-gal activity (units =1000 × A420/ t × 5 × A600) calculation, the experiment was repeated three times to take the mean. Positive control, P53 + T; Negative control, Lam + T. ap < 0.05. B: Immunoprecipitation assay. HEK293 cells were transfected with plasmid pcDNA3-C9, then infected with HSV-1 (MOI = 20). Cells were harvested for immunoprecipitation with C9 or UL25 antiserum. C: Detection of co-localization by dual-fluorescence. The localization in Vero cells of fusion proteins pEGFP-C9 (green fluorescent) and pDsRed-UL25 (red fluorescent). Negative control, pEGFPN2. Bar, 10μm
Cellular function of C9orf69
A preliminary analysis of C9orf69 suggests that the protein is only expressed in the course of HSV-1 infection or at a special time (Fig. 3A). Therefore, up-regulation of the protein may be a cellular response to specific circumstances. Preliminary analysis using MTT indicated that compared to the control, there was a significant increase in cell proliferation when the C9orf69 expression plasmid was transfected into cells (Fig. 3B). The cyclic distribution of these cells as seen by flow cytometry suggested that C9orf69 expression in cells significantly promotes progression of cells into S phase as compared to controls (Fig. 3C). These results indicate that C9orf69 plays a role in accelerating cell proliferation by promoting progression into S phase and maintaining cells in S phase. However, the mechanism still needs to be studied further.

Figure 3. Possible biological function of C9orf69 up-regulation in cells. A: Reactive generation of C9orf69. HEK293 cells were infected with HSV-1 (MOI = 0.01) and RNA was extracted at different times to amplify C9orf69 mRNA by real-time qPCR. Using β-actin mRNA as internal reference standards, relative quantitative processing was done by lg2-⊿⊿t. The expression of C9orf69 in HEK293 cells not infected with HSV-1 is 1. B: Impact on cells by C9orf69 with MTT assay. HEK293 cells were transfected with pcDNA3 and pcDNA3-C9 respectively every 12 h for the MTT assay. C: Detection of C9orf69 influence on the cell cycle by flow cytometry. HEK293 cells were transfected with pEGFPN2 and pEGFP-C9 and samples were collected at different times for cell cycle analysis. ap < 0.05; bp < 0.01
Increase of HSV-1 proliferation efficiency by C9orf69
Our studies of HSV-1 growth after up-regulating C9orf69 showed that compared to controls, C9orf69 significantly increased HSV-1 proliferation efficiency in infected cells (Fig. 4A). Peak viral proliferation and infection advanced 10 h earlier than the control, which means that C9orf69 protein may promote the growth of virus at some stage in the course of HSV-1 proliferation. On the contrary, the speed of viral growth was slower in C9orf69 silenced cells (Fig. 4B). The results of plaque assay showed a similar tendency in the change of viral load (data not shown). An analysis of current data on HSV-1 proliferation suggests that the speed and efficiency of viral multiplication is based mainly on the transcription and assembly of the viral genome and structure [10]. On the basis of these data, a preliminary study of promoter function in the representative genes (ICP4 (immediate-early coding protein 4), TK (thymidine kinase), gC (glycoprotein C) of HSV-1 α, β and γ genes) potentially regulated by C9orf69 was performed using the CAT (chloramphenicol acetyl transferase) assay[21]. The results suggest that C9orf69 protein has no direct effect on transcription of these three types of viral genes (data not shown here). Because the target molecule of C9orf69 is a viral structural protein, translocation of the protein was observed in the course of HSV-1 infection using cells transfected with fluorescent pEGFP-C9. The results showed that under noninfectious conditions, the location of C9orf69 protein displayed mid-point distribution in the cytoplasm (Fig. 5), but in HSV-1 infected cells, its localization was more dynamic. At first, within 6 h of viral infection, the C9orf69 tended to accumulate in the cytoplasm and around the nuclear membrane (Fig. 5). Then, the protein spread further into the cytoplasm with a small population entering the nucleus (Fig. 5). The results indicated that C9orf69 protein might participate in the transfer of some viral components in the course of a HSV-1 infection. Combined with the finding that C9orf69 can interact with UL25, it can be speculated that C9orf69 may be involved in the viral assembly process associated with UL25 and can promote viral proliferation.
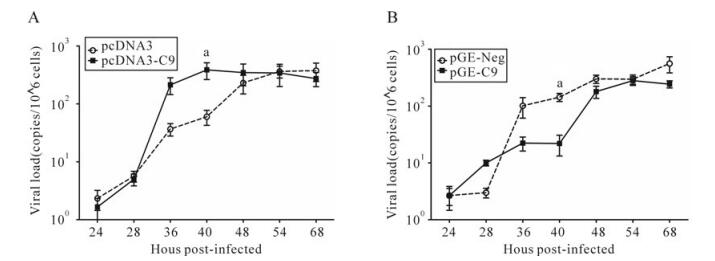
Figure 4. C9orf69 promotes HSV-1 proliferation. A: HEK293 cells were transfected with pcDNA3 and pcDNA3-C9. B: HEK293 cells were transfected with pGE-Neg and pGE-C9. 10 h after transfection of infectious HSV-1 (MOI = 0.01), samples were collected at different times, viral DNA extracted and the UL35 gene was amplified by real-time PCR. ap < 0.05
-
Like most viruses, due to the limited number of genes carried, herpes virus requires resources provided by its external host environment for growth and proliferation [9]. On the other hand, the host has developed a series of natural and acquired immune mechanisms to resist foreign organisms from invading [5]. Regardless, both involve mutual interaction of viral and host molecules.
As a complex DNA virus, HSV-1 may influence cellular protein expression through its interaction with host cells after infection [16, 20]. These changes in protein level alter the microenvironment in the cell and make HSV-1 enter into a latent or cleavage infection process [16]. Cell proteins induced by viral infection include proteins related to natural immunity [7] as well as proteins that play a main role in promoting viral growth and proliferation [4]. The host protein C9orf69 is an example of a protein produced in response to HSV-1 infection (Fig. 3A).
The results provided in our study show that C9orf69 may promote HSV-1 proliferation efficiency (Fig. 4), but the mechanism does not involve transcription regulation or replication of viral genes. C9orf69 is mainly localized in the cytoplasm, and it is widely distributed in the cytoplasm even after HSV-1 infection (Fig. 5). Theoretically, there is no possibility for the protein to participate in transcription and regulation of genes. Experiments in vitro demons-trated that C9orf69 could interact with the structural protein UL25 associated with viral transport and assembly (Fig. 2), suggesting that the function of C9orf69 may be closely related to its interaction with UL25. In fluorescence experiments, C9orf69 localization in cells varied dynamically with the progression of viral infection (Fig. 5). However, most molecules were moving around the cytoplasm or nuclear membrane with only a small amount localized in the nucleus. These results suggest that C9orf69 may function through non-transcriptional mechanisms such as transport or assembly, to exert its influence on viral growth and proliferation.
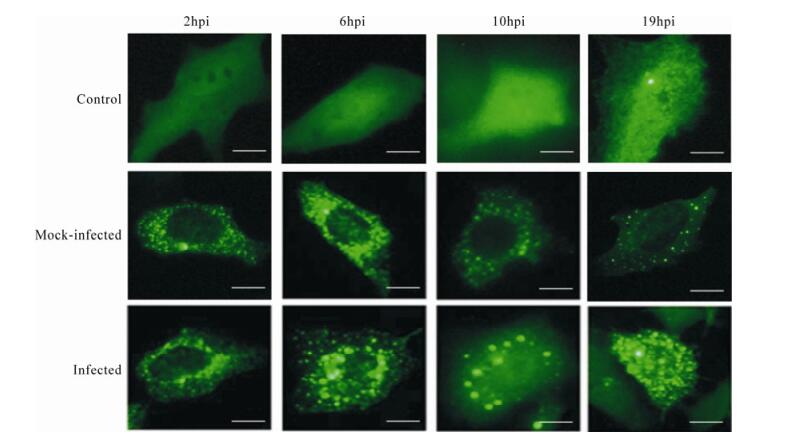
Figure 5. Changes in C9orf69 localization in cells after HSV-1 infection. HEK293 cells were transfected with pEGFP-C9, then infected with HSV-1 (MOI = 0.01) 24 h after transfection. Samples were collected at different times for fluorescence microscopy. Control (transfected pEGFPN2), mock-infected (transfected with pEGFP-C9 but not infected) and infected (transfected with pEGFP-C9 and infected) in HEK293 cells. Bar, 8μm
Our experiments also show that as a reactive protein, C9orf69 not only affects proliferation of HSV-1 (Fig. 4), but also the proliferation of cells (Fig. 3B and 3C). This may explain why C9orf69 expression displays very low expression in normal cells. However, HSV-1 infection of cells may gradually change the microenvironment of host cells, affecting the cell cycle. Studies have shown that viral proteins ICP0 can stop the host cell cycle at the G1/S phase [3, 14, 15], which is conducive to early viral protein expression; ICP22 changes the cell cycle, which is more suitable for viral DNA replication and late protein expression [1]. Flow cytometry experiments confirmed that C9orf69 could change the cell cycle and affect cell growth by an unknown mechanism (Fig. 3C). The interaction of the UL25 protein of HSV-1 with C9orf69 might indirectly affect cellular activities. Therefore, HSV-1 may utilize this mechanism to alter the cell environment to meet its own needs.
It is assumed, based on the above results, that C9orf69 is induced by HSV-1 infection of cells and can interact with viral UL25 protein and jointly translocate to the nucleus. This accelerates viral DNA release in the nucleus, allowing the virus to initiate its replication and proliferation processes.
Acknowledgments
This work was supported by Scientific Research Fund of the Institute of Pathogen Biology (No. 2007IPB10).

 DownLoad:
DownLoad: