











-
Ever since the pioneering work of the Khorana' group that synthesized the 77-bp nucleotide gene encoding a yeast alanine transfer RNA and the 207-bp gene for the tyrosine suppressor tRNA[1, 19] 40 years ago, scientists have had the ability to join DNA sequences and produce combinations that are not present in nature. Recently, chemical synthesis of genes has become routine, and the chemical synthesis of genomes has now become a reality and several viral genomes generated by the chemical synthesis have been reported[4, 20]. Gibson et al. developed a method for assembling the 1.08-mega-base Mycoplasma mycoides genome staring from digitized genome sequence information, and successfully transplanted it into a Mycoplasma capricolum recipient cell to create a modified Mycoplasma mycoides cell controlled by a synthetic genome [6].
Transfusion Transmitted Virus was first reported as a post-transfusion hepatitis virus of unexplained etiology in Japanese patients in 1997[14, 15] and was recently named as Torque teno virus (TTV) as the first member of the new family Anelloviridae. The virus is a nonenveloped virus containing a single-stranded, circular DNA genome of approximately 3.8 kb [13, 22]. Until now, at least 39 genotypes of TTV have been identified, and they can be classified into five distantly related groups [18]. The TTV genome includes an untranslated region (UTR) of approximately 1.2kb and a coding region of approximately 2.6kb, including two main open reading frames (ORFs) which are sandwiched by the TATA box and polyadenylation signal motifs[13, 14, 16]. Epidemiological studies have shown that TTV is genetically variable and widespread in the general human population[2]. Although replication of TTV indicates that it occurs in the liver [17], and most researchers have considered that it might have a possible association with non-A-G hepatitis, post-transfusion hepatitis which may induce liver disease [3, 10, 15, 16], to date there is no consistent evidence of a link between TTV infection of humans and specific disease [11]. The virus is extremely common, even in healthy individuals-as much as 100% prevalent in some countries, and in approximately 10% of blood donors in the UK and the US. The exact role of this virus in the pathogenesis of chronic liver diseases remains controversial.
In this study, we obtained the entire 3.8 kb synthetic TTV genome using a purely in vitro process. Through this work, some experience in assembling the genomes containing complex structures from oligonucleotides has been gained, thereby we can reconstruct viral genomes less than 10 kb and accelerate the preparation of live attenuated vaccines. Viruses also represent natural systems which can deliver foreign genes into target cells; therefore, the artificial TTV genome could be developed as a hepatic disease gene therapy vector in the future.
HTML
-
PCA and SOEing methods
PCA (polymerase chain assembly reaction) is a method of assembling many oligonucleotides into a longer double-strand DNA employing thermostable polymerase [5]. We used PCA to obtain assembled cassettes from chemically synthetic oligonucleotides and then added a subsequent PCR step based on the two terminal oligos to amplify the assembled product (Fig. 1). SOEing (splicing by overlap extension) is a method for joining two PCR products into one with short homologous sequence [9]. We used SOEing to obtain assembled earlier intermediates and the full length viral genome from cassettes and latter intermediates. In our experiment, we used more than two PCR products as the templates, let them extend with partial overlaps anneal at the first reaction and then added a subsequent PCR based on the two terminal oligos to amplify the full length product (Fig. 1).
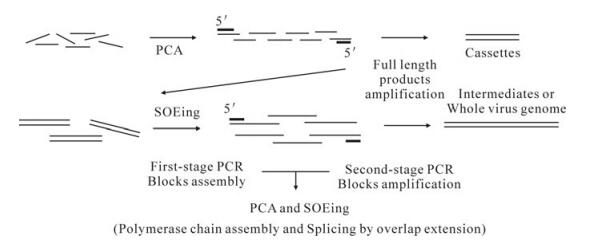
Figure 1. Two methods for assembly of oligos or cassettes. Synthetic oligonucletides are mainly assembled by PCA, cassettes and intermediates are mainly assembled by SOEing
Synthetic oligonucleotides design
TTV (SANBAN isolate) genome (GenBank accession number AB025946) [23] was divided into 102 overlapping oligos. These coupled primers were named F1/R1, F2/R2, …, F51/R51. The chemical synthesis oligonucleotides were generally 55-60 base oligos with 15-20 bases overlaps to adjacent oligos and purchased from Sangon Biotech (Shanghai) with ULTRAPAGE purification. The stock solutions were prepared at a concentration of 100 μmol/L in Tris/EDTA (TE) buffer, pH 8.0.
Preparation and assembly of ~400 bp cassettes
Oligonucleotides were assembled by PCA method. PCA was carried out in 25 μL reaction mixtures containing 400 nmol/L each of the 8 or 10 oligos (4 or 5 coupled primers), 200 μmol/L each dNTP, 1.2 mmol/L MgSO4, 0.5 μL KOD-PLUS DNA polymerase (TOYOBO) and 2.5 μL 10×PCR buffer for KOD-PLUS. The assembled program consisted of 35 cycles of 94℃ for 30 s, 45℃ for 30 s, and 68℃ for 30 s.
The assembled cassettes produced by PCA were amplified. The 25 μL PCR mixtures contained 1 μL PCA product (unpurified), and 1.5 μL each 10 μmol/L two terminal oligos, 200 μmol/L each dNTP, 1.2 mmol/L MgSO4, 0.5 μL KOD-PLUS DNA poly-merase (TOYOBO) and 2.5 μL 10×PCR buffer for KOD-PLUS. PCR parameters were 94℃ for 2 min, then 32 cycles of 94℃ for 30s, 50℃ for 30 s, and 68℃ for 30s, followed by 68℃ incubation for 5 min. The 75 μL(25 μL×3) PCR amplification products were purified using the Gel Extraction Kit (OMEGA) according to the provided manual, and re-dissolved in 25 μL of steriled ddH2O.
Preparation and assembly of GC-rich and hairpin structure cassettes
The TTV (SANBAN isolate) genome has a GC-rich region located at nucleotides 3697-3808 and a stable hairpin structure at nucleotides 3153-3251[8], thus we couldn't assemble the short original oligonucleotides that contained these secondary sequences by PCA. Therefore, we designed a 150 base (R53) and a 128 base (R42-43) single-stranded DNA oligos with 15-20 bases overlaps to adjacent oligos that contained the full-length GC-rich sequence and the hairpin structure sequence respectively, (purchased from Integrated DNA Technologies, Inc (Coralville, Iowa, USA) with standard desalting purification) and dissolved them in 100 μmol/L Tris/EDTA (TE) buffer, pH 8.0.
The 150 base oligos (R53) and a 128 base oligos (R42-43) had the following sequences: 5'-GTCACG TGGTTAGTGACGGACTTCGGGGGGGGGGCCGGGGGGCAAACCCCCCCCCGGGGGGGGCCCCCCCCTTTCCCCGGGGGGGGCGGCAGCCCCCCGGCGCGCGCGCGCAGCGCGCGCGCCGCCGCCGAGCCGTTATTTTTAAAAAAG-3' and 5'-TCGGA GTCTGTTTAGCAAAATTCCCGACCCCCGCGAGCCCGAAGTGGTCTGACACGCGCGAGCGTGTC AGCACGAGCGGGGGTCTGAGGTGCCGCGCGCAGCCGAAGGCGTAGCGCGCGGCTCCGAAG-3'.
We set up 25 μL PCA reactions using 400 nmol/L each of the 4 or 6 oligos, 400 μmol/L each dNTP, 0.5 μL KOD-FX DNA polymerase (TOYOBO) and 12.5 μL 2×PCR buffer for KOD-FX. PCR parameters were 94℃ for 2 min, then 32 cycles of 98℃ for 15 s, 58℃ for 30 s, and 68℃ for 1 min, followed by 68℃ incubation for 5 min.
We amplified the PCA products with two terminal oligos as primers. The 25 μL PCR mixtures contained 1 μL PCA product (unpurified), and 1.5 μL each 10μmol/L forward and reverse oligos, 400 μmol/L each dNTP, 0.5 μL KOD-FX DNA polymerase (TOYOBO) and 12.5 μL 2×PCR buffer for KOD-FX. PCR parameters were 94℃ for 2 min, then 28 cycles of 98℃ for 15 s, 60℃ for 30 s, and 68℃ for 1 min, followed by 68℃ incubation for 5 min. The PCR products were purified and re-dissolved as above.
DNA cloning and sequencing
The synthetic DNA fragments were purified by gel extraction and integrated into the vector pTA2 (TOYOBO) using the TA cloning System. The ligation products were transformed into DH5 E. coli cells and selected on LB plates with 50 μg/mL ampicillin, 20 μL 100 mmol/L IPTG and 4% X-gal. Individual E. coli colonies were picked and incubated overnight in 1.0 mL LB liquid medium with 50 μg/mL ampicillin at 37℃.
To screen for full-length first-stage intermediates, we performed PCR of cultured bacteria. The bacteria were used as templates in a PCR reaction with the primers complementary to the terminal sequences of synthetic DNA fragments. We set up 10 μL reactions using 0.4 μL template, 400 nmol/L each primer, 400 μmol/L each dNTP, 0.2 μL KOD-FX DNA polymerase (TOYOBO) and 5 μL 2×PCR buffer for KOD-FX. Cycling parameters were 94℃ for 2 min, then 32 cycles of 98℃ for 15s, 55℃ for 30 s, and 68℃ 1 kb per 1 min of amplicon, followed by 68℃ incubation for 5 min. We analyzed products on 1% agarose gel alongside the DNA ladder. Plasmids containing the fragments of interest were extracted from these cells using the E.Z.N.A.TM Plasmid Mini Kit (OMEGA) and sequenced.
Preparation and assembly of ~800 bp intermediates
We used SOEing to assemble synthetic cassettes produced by PCA.The PCR reaction mixtures (25 μL) contained 1μL each of 2-3 purified 400 bp cassettes, 400 μmol/L each dNTP, 0.5 μL KOD-FX DNA polymerase (TOYOBO) and 12.5 μL 2×PCR buffer for KOD-FX. Cycling parameters were 94℃ for 2 min, then 30 cycles of 98℃ for 15 s, 58℃ for 30 s, and 68℃ 1 kb per 1 min, followed by single 68℃ incubation for 5 min.
The synthetic intermediates produced by SOEing were amplified. The 25 μL PCR mixtures contained 1 μL unpurified SOEing product, and 1.5 μL each 10 μmol/L forward and reverse primers, 400 μmol/L each dNTP, 0.5 μL KOD-FX DNA polymerase (TOYOBO) and 12.5 μL 2×PCR buffer for KOD-FX. PCR parameters were 94℃ for 2 min, then 28 cycles of 98℃ for 15s, 60℃ for 30 s, and 68℃ 1kb per 1min, followed by single 68℃ incubation for 5 min. The PCR products were purified, cloned and sequenced as above.
Genome assembly and amplification
After sequencing, the 2 849 bp, 368 bp and 805 bp verified synthetic intermediates produced by SOEing were amplified and purified as above. We also used SOEing to assembly the synthetic intermediates into the entire TTV genome. The reaction condition was similar to 800 bp intermediates assembly, while the annealing temperature (Ta) was raised by 2℃ in each reaction. The PCR amplification products were purified, cloned and sequenced as above.
-
As many as 5 overlapping oligo pairs are easily assembled into 400 bp cassette at once
The native 3 808-bp TTV (SANBAN isolate) genome (GenBank accession number AB025946)[23] was divided into 102 overlapping 55-60 base oligos (coupled primers F1/R1, F2/R2, …, F51/R51). Two single-stranded DNA oligos with 15-20 base overlapping could be assembled to 90-100 bp ds-DNA.
To determine the capacity for oligo assembly in one PCA reaction, we took eight, five, three, two and two oligo pairs as templates in each of five individual PCA reactions. The gel electrophoresis' results showed that, in the first stage of PCA assembly, only a small fraction of full-length assembly product was visible when mixing two or three oligo pairs in reaction mixtures. After second-stage amplification, there were specific assembly bands observed when the oligo pairs were not more than five (Fig. 2). This provided evidence that as many as 5 overlapping oligo pairs are easily assembled into 400 bp cassette at once under our reaction conditions.
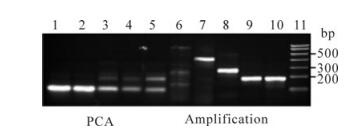
Figure 2. Gel electrophoresis' analyses of assembly of oligonu-cleotides by two-steps PCA at once. 1-5: Oligonucletides are assembled by PCA assembly reactions; 6-10: PCA assembled products are amplified by PCR. Lane 1, Eight oligo pairs; 2, Five oligo pairs; 3, Three oligo pairs; 4, Two oligo pairs; 5, Two oligo pairs; 6, Eight oligo pairs; 7, Five oligo pairs; 8, Three oligo pairs; 9, Two oligo pairs; 10, Two oligo pairs; 11, DNA Marker I from TransGen Biotech(Beijing)
Assembly of the TTV genome nucleotides 78-2926 sequence
To assemble the TTV genome 78-2 926 sequence with oligonucleotides, the 55-60 base oligos were assembled in groups of four or five pairs into eleven 350-400 bp cassettes. These were joined in sets of two or three to produce four assemblies of approximately 800 bp and then again in sets of two to produce two 1.5 kb assemblies. These two fragments were recombined into approximately 3.0 kb (2 849 bp) synthetic intermediates (Fig. 3A). The assembled intermediate 02-38 (the 78-2926 sequence) was detected by agarose gel electrophoresis, the result showed that we obtained the specific positive band, though there were some non-specific bands under 1.0 kb (Fig. 3B). The synthetic DNA fragments with expected length were Gel extracted and sequenced.
Figure 3. Assembly of TTV genome nucleotides 78-2926 sequence (oligo pairs F2/R2-F38/R38). A: Strategies for the sequence synthesis. B: Gel electrophoresis' analyses of synthetic assembled intermediate. Lane 1, As many as 37 oligo pairs were assembled into intermediate 02-38(the 78-2926 sequence); 2, 1kb DNA marker from Fermentas (Burlington, Ontario, Canada)
Assembly of the GC-rich (3612-3808 and 1-171) and the hairpin structure (2842-3646) sequences
We designed a 150 base (R53) single-stranded DNA oligo and a 128 base (R42-43) single-stranded DNA oligo with 15-20 bases overlaps to adjacent oligos which contained the full-length GC-rich sequence and the hairpin structure sequence respectively, and assembled them with PCA into two cassettes. GC-rich and the hairpin structure cassettes of the correct sequence were assembled to produce 368 bp and 805 bp intermediates individually (Fig. 4A, 4C). The assembled intermediates 49-02 (3612-3808 and 1-171 sequence) and 38-49 (2842-3646 sequence) were detected by agarose gel electrophoresis, the result showed that we obtained a positive band of the estimated size (Fig. 4B, 4D). The synthetic DNA fragments with expected length (368 or 805 bp) were cloned and sequenced.
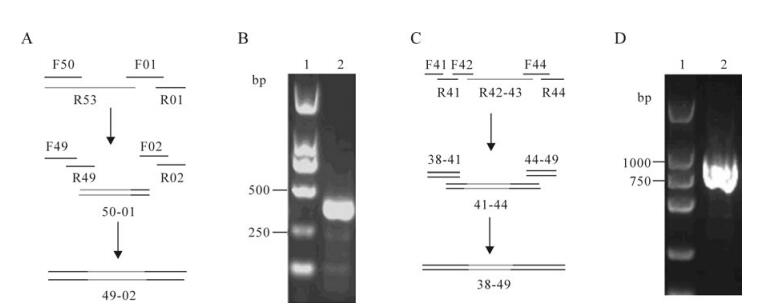
Figure 4. The GC-rich and the hairpin-structure sequences synthesis. A: Strategies for the GC-rich sequences (3612-3808 and 1-171) synthesis. B: Gel electrophoresis' analyses of the GC-rich sequences assembly reactions. Lane 1, Marker I from Dongsheng Biotech (Guangzhou); 2, As many as 4 oligo pairs (from F49/R49 to F02/R02) were assembled into intermediate 49-02(368bp). C: Strategies for the hairpin-structure sequences (2 842-3 646) synthesis. D: Gel electrophoresis' analyses of the hairpin-structure sequence assembly reactions. Lane 1, Marker I from Dongsheng Biotech; 2, As many as 11 ologo pairs (from F41/R41 to F49/R49) were assembled into intermediate 38-49(805bp)
Assembly of 3.8 kb TTV full length genome
The larger intermediates 02-38, 38-49, 49-02 were cloned in the pTA2 vector(TOYOBO) and verified by sequencing, then, these three fragments were assembled into a complete TTV genome (Fig. 5A). Gel electro-phoresis' results showed that a specific band of approximately 3.8 kb matched the expected size (Fig. 5B). The synthetic genome were cloned and sequenced. The 3.8 kb synthetic genome was successfully aligned to natural full length genome sequences using the online ClustalW Multiple alignment tool (http://www.ebi.ac.uk/Tools/msa/clustalw2/) indicating we successfully synthesized 3.8 kb TTV full length genome.
Figure 5. Strategy for the TTV genome synthesis and analyses. A: Strategy for the 3.8 kb TTV (SANBAN isolate) genome synthesis. B: Gel electrophoresis' analyses of entire synthetic TTV genome synthesis reactions. Lane 1, 5kb marker from TAKARA BIO (Otsu, Shiga, Japan); 2, The 3 intermediates (02-38, 38-49 and 49-02) were assembled into entire synthetic 3.8 kb TTV genome
-
Oligonucleotides assembly by PCA and double-strand DNA connection by SOEing, as described here, could be very useful in the synthesis of DNA fragments, including genes and genomes, less than 10kb. In fact, the principles behind both methods is similar, as they both require two-steps. On the first step, the templates are presented in approximately the same quantities will anneal with partial overlaps and extend, so that each becomes longer extensions. Then, the extension products can be amplified on the second reaction step.
The assembly of a viral genome from synthetic oligos could be performed according to the protocols we have described here. The length of the oligos used for assembly is an important factor that influences error rates of the final products of DNA synthesis. There are fewer carry-over errors when shorter oligos are used, but total producing cost is more expensive. Xiong et al. considered that 60-base oligos provided a reasonable balance between low error rate and low cost [24], so we followed the principle in this study.
The limits of oligos uptake and assembly have been explored. Stemmer et al. had assembled a 2.7 kb plasmid used 134 oligos in a single reaction[21]. But in our single reaction, the capacity to efficiently assemble into full-length cassettes was not more than five oligo pairs, and only approximately 400 bp assemblies were produced. In Stemmer's experiment, the assembled cycles of PCA reaction reached 55 and the working concentration of each oligo was less than 50 nM. In the future, the oligo uptake limits in our experiment can probably be improved with increasing the numbers of assembled cycles and diluting the oligo concentration.
In most genomes of organisms, there are some gene-regulatory elements, including promoters, enhancers and other control elements, consisting of GC-rich regions [7]. Assemblies of these regions are essential in synthesis of entire genomes to maintain vital functions. DNA regions with GC-rich content are more prone to form secondary structures with high melting temperatures (Tm). In the case of GC-rich sequences, the single-stranded template may form intra-molecular stem loops during the initial cycles of amplification, which can stall the reaction. So, the longer oligos must be synthesized containing the whole-length GC-rich region, while homologous flanking sequences with low GC-content are also required. Nowadays oligos as long as 150-200 bp can be chemically synthesized by several biotechnological company, such as Integrated DNA Technologies. In order to improve the amplification efficiency of GC-rich targets, we utilized KOD-FX (TOYOBO) in the PCA reaction to assemble the GC-rich and the hairpin structure sequences, owing to its high fidelity, excellent elongation ability and ability to amplify of GC-rich target and crude samples. Furthermore, the annealing temperature (Ta) used during this reaction needed to reach at least 60℃.
TTV genome is a single-stranded circular DNA, therefore, cyclization and revival of entire synthetic genome was carried out in our laboratory. This study demonstrates that entire viral genomes and synthons can be effectively assembled by the methods described here and provides a methodological basis for chemical synthesis of a viral genome for used as a live attenuated vaccine or gene therapy vector.
Here we have successfully synthesized the 3.8 kb TTV genome containing a hairpin structure and a GC-rich region. However, such in vitro synthesis methods are adequate for generating DNAs up to several tens of kb in length. Application of synthetic technology in vitro and following Gibson's transformation-associated recombination (TAR) method in yeast [6], much larger genomes could be synthesized. We plan to design, assemble and clone genomes of cyanophage, such as Pf-WMP3 of 43 kb genome and Ma-LMM01 of 162 kb genome, which infect cyanobacterium Phormidium foveolarum and Microcystis aeruginosa respectively[12, 25]. The synthetic cyanophage could serve as potential algicide candidate against toxic cyanobacterium to control water bloom.
Acknowledgements
We thank Dr. Zhengli Shi, Dr. Zhihong Hu, Dr. Xinwen Chen and Dr. Simon Rayner of Wuhan Institute of Virology, Chinese Academy of Sciences, China, for their advice, encouragement and support in conducting this research project. This work was supported by the Knowledge Innovation Program of the Chinese Academy of Sciences, Grant No. KSCX2-EW-Z-3.

 DownLoad:
DownLoad: