











-
BTI-Tn5B1-4 (High5) cells, as well as IPLB-SF21 and its clone (SF9), are the most widely used insect cell lines for baculovirus expression vector systems [5, 6, 8]. The maintenance and growth of these and other insect cell lines need particular attention since the characteristics of cells may change upon long-term passaging [1, 4, 15, 16]. In many instances, High5 cells provide superior production of recombinant proteins when compared to SF9 cells; this high productivity is more evident in low passage cells [15]. Donaldson and Shuler reported that early passage High5 cells (passage 130) expressed more recombinant secreted alkaline phosphatase (SEAP) than high passage cells (passage 360) [4]. We also got the same results using two clones designated as H5CL-B and H5CL-F from low passage High5 cells (passage 90). Both clones were distinct from each other or the parental cells in terms of morphology and expression of recombinant proteins but exhibited a similar growth rate in serum-containing TNM-FH medium. Both H5CL-B and H5CL-F clones produced 30% and 45% more β-galactosidase than the parental High5 cells (passage 161). Similarly, both clones produced 100% more secreted alkaline phosphatase than the parental cells [9, 15]. Recently however, an adventitious virus was identified in High5 cell lines. Latent infection by the alphanodavirus, TNCL virus (Tn5 Cell line virus), was reported during production of hepatitis E virus-like particles in High Five cells infected with a recombinant baculovirus vector [10]. Soon afterwards, we examined H5CL-B and H5CL-F cells for the presence of the TNCL virus. The TNCL virus was found in both H5CL-B and H5CL-F cells, too. Therefore, we developed a novel cell clone from a new cell line which has been confirmed to be free of TNCL virus [12]. Here we describe the generation and the characteristics of the clone.
HTML
-
High Five and SF9 Cells were used as positive and negative controls in this study, respectively. Both the cell lines were obtained from Dr. Granados [6]. Cell line QB-Tn9-4s was established by Dr.Li's laboratory [12]. All of viruses were obtained from Dr. Ping Wang, which included: a) wild type (WT) AcMNPV [12], b) a recombinant AcMNPV-β-gal. (containing the Eschericha coli β-galactosidase gene under the transcriptional control of the polyhedron gene promoter) [15], and c) a recombinant AcMNPV-SEAP (containing a secreted human placental alkaline phosphatase gene under the control of a polyhedron promoter) [2].
-
In order to determine if the parental cells, QB-Tn9-4s, carried the TNCL virus, we used RT-PCR to examine AcMNPV infected cells and uninfected cells from QB-Tn9-4s, High Five and SF9 cells were used in this study because the virus was shown to persistently infect cultures of High Five cells, and is produced in High Five cells at a significant level when challenged by wild type or recombinant AcMNPV [10]. To detect TNCL virus four sets of primers, which cover the bipartite TNCL genome segments (1 and 2), were designed (table 1) and used for one-step RT-PCR analysis of total RNA from QB-Tn9-4s, High Five and SF9 cells that were infected with AcMNPV at MOI of 10. Total RNA was extracted from infected cells at 48 h p.i. using Trizol reagent (Invitrogen) according to the manufacturer's protocol. These four primer sets were used to sample the bipartite RNA genome of the TNCL virus. The four primer sets produced amplicons with the expected sizes from AcMNPV-infected High Five cell RNA (table 1). Each PCR consisted of 25 μL reaction mixture containing 1 × RT-PCR buffer (Invitrogen, 12.5 μL), forward primer (10 μm, 1μL), reverse primer (10 μm, 1 μL), SuperScript Ⅲ (SS Ⅲ) enzyme mixture (0.5 μL), RNase free water (9 μL), and purified RNA (0.5 μg/mL, 1 μL). RT-PCR was run under the following condition: 1 cycle of 45℃, 30 min; 1 cycle of 95℃, 3 min; 35 cycles of 94℃, 20sec/ 54℃, 20 sec/72℃, 1 min. RT-PCR products were separated on a 1% agarose gel.

Table 1. Primers and RNA sequences used in TNCL virus study
-
QAU-BTI-Tn9-4s cells (QB-Tn9-4s) at passage 91 were used for the cell cloning [12]. The cell cloning was carried out with a 24-well cell cloning plate (Greiner Labtechnik), that consists of 16 grids (each 2mm square) per well [9]. Briefly, 2mL of cell suspension by limited dilution was placed in the well and the cells were incubated for 2h attachment. All of the single cells in each grid were marked under a microscope (Olympus IX71) then the plate was incubated at 28℃. The cells were transferred into 96-well plates when the cells were confluent on the bottom of the grids. Finally, the cells were transferred into a tissue culture flask. Among thirty cell clones cloned as single cells, eight rapidly growing cell clones were selected for further screening and evaluation. The screening and evaluation consisted of cell selection for rapid growth characteristics, high susceptibility to Autographa califomica nuclear polyhedrosis virus (AcMNPV) infection, and high expression of recombinant proteins. Finally, two clones, QB-CL-A and QB-CL-B, were obtained for further comparative analysis with the parental QB-Tn9-4s cells and BTI-Tn5B1-4 (High Five) cells.
-
Microscopic images of cells from individual cell lines were obtained by using an Olympus IX 71 inverted phase contrast microscope and printed. From these prints, cell size was measured and converted to actual size according to a calibrated manufacturing factor. Average cell dimensions were determined from measurement of 50 cells [14].
Triplicate 25cm2 flasks of QB-CL-B, QB-CL-A, QB-Tn9-4s, and Tn5B1-4 in log phase were seeded at a density of 1.0 × 106 cells/flask each and allowed to grow at 28℃. At 24 h intervals the cell densities and viability from replicate flasks were determined by using a hemacytometer and trypan blue staining. Cell population doubling times were calculated using the exponential formula described by Hayflick [7] and cell growth curves were generated from the results of three independent experiments.
-
Tn5B1-4 (attached cells) cells in log phase were seeded in triplicate into 24-well plates (Corning, USA) at a density of 1.0 × 105 cells/well and allowed to attach for 2 h at 28℃. Following gentle removal of the spent medium, 0.5 mL AcMNPV-1A budded virus (BV), multiplicity of infection [MOI=10] was added into each well and incubated for absorption for 1 h on a shaker. After gently removing the inoculum and washing with fresh medium, 2 mL of fresh medium was replaced. For QB-CL-B, QB-CL-A, and QB-Tn9-4s (suspended cells), the cells were incubated with AcMNPV infectious medium in suspension at an MOI of 10 at 28℃ culture for 2 h on shaker. After centrifugation at 1500 r/min for 5 min, the cells were washed then suspended in fresh medium to a cell density of 0.5 × 105 cells/mL. Following this, aliquots of 2mL of the cell suspension were placed into individual wells. All of the plates were incubated at 28℃. The culture medium was collected to determine the production of BV using virus titration by TCID50 on SF9 cells then the ratio of infection at 4 days post-infection (p.i.) was calculated before the OBs from cells were released. After the cells were disrupted by sonication to release virus occlusion bodies (OBs), the number of OBs from each cell were calculated as previously described by Wang et al. [13].
-
To compare recombinant protein production in the cell lines, QB-CL-B, QB-CL-A, High Five, and QB-Tn9-4s cells were infected with a recombinant virus (β-gal-AcMNPV) expressing the intracellular protein β-galactosidase, or a recombinant virus (SEAP-AcMNPV) expressing the secreted protein, SEAP. The suspended cells (QB-CL-B, QB-CL-A, and QB-Tn9-4s) and attached cells (High Five) were infected by different methods [11]. The infection procedures were carried out in the same manner as that for study of AcMNPV virus yields (as described above).
For β-galactosidase, both the infected cells and medium were collected by gently scraping cells from plates and pelleting at 10000 r/min for 1 min. The cells were sonicated for production of lysates then centrifuged at 12000 r/min for 5 min. 2 μL of supernatant was added to 80 μL of Z-buffer (60 mmol/L Na2HPO4, 40 mmol/L NaH2PO4, 10 mmol/L KCL, 1 mmol/L MgSO4, 50 mmol/L β-mercap-toethanol, pH 7.4) in a 96-well plate. 20 μL of pre-warmed substrate solution(OPNG, 4 mg/mL in Z-buffer)was added to start the reaction. After 2 min of incubation at 28℃, 0.5 mL of 1 mol/L Na2CO3 was added to stop the reaction. β-galactosidase activity was monitored by readings at 420 nm (Microplate photometer, MRXII) and converted to enzyme activity in IU/mL as described by Wang et al. [13].
In order to assay for SEAP, infection with SEAP-AcMNPV was carried out using the same method described above. Endogenous alkaline phosphatase activity present in the infected culture medium was inactivated by incubation of supernatant sample at 65℃ for 5 min. 2 μL of each infected cell supernatant sample (containing SEAP) was mixed with 200μL of SEAP buffer (1.0 mol/L diethanolamine, pH 9.8, 0.5 mmol/L MgCL2, 10 mmol/L homoarginine) in a well of a 96-well plate, and the mixture was incubated at 27℃ for 30 min. 20 μL of freshly prepared and pre-warmed substrate solution (40 mmol/L P-nitrophenylphosphate in SEAP assay buffer) was added to each well. Absorbance at 405 nm was monitored by a previously described protocol [3, 13]. The SEAP production was calculated according to the formula by used Meng et al. [12].
Materials
Analysis of TNCL Virus
Cell Cloning and Selection
Cell Morphological Observation and Growth Curves
AcMNPV Infectivity and Production of Virus
Analysis of Recombinant Proteins Production from Recombinant AcMNPV
-
Recently, an alphanodavirus (TNCL Virus) was described in High Five cells (Tn5B1-4). TNCL virus was induced upon infection by wild and recombinant AcMNPV. In order to determine whether QB-Tn9-4s cells also carried this virus, we used RT-PCR to examine AcMNPV infected and uninfected cells from QB-Tn9-4s, SF9, and High Five. High Five and SF9 were used as positive and negative controls, respectively. Four primer sets were used to sample the bipartite RNA genome of TNCL virus (Table 1, Fig. 1). The four primer sets produced amplicons with the expected sizes from AcMNPV infected High Five cell RNA, confirming the presence of TNCL virus. However, in a parallel analysis no amplicons were detected from cellular RNA preparations from AcMNPV infected QB-Tn9-4s and SF9 cells (Fig. 1). Based on the results that the parental cells, QB-Tn9-4s, were free of the TNCL virus, we finally decided to carry out cell cloning from QB-9-4s cells to obtain a novel cell clone, QB-CL-B, with high expression of both β-galactosidase and SEAP.
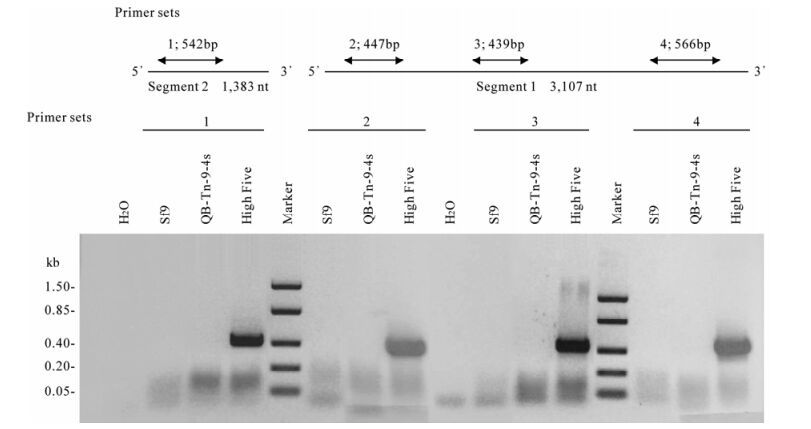
Figure 1. RT-PCR analysis and TNCL virus detection. To examine cell lines for the presence of a TNCL virus, four primer sets were used for RT-PCR analysis of total RNA from cells. SF9, QB-Th9-4s, and High Five cells were infected with AcMNPV 48 h prior to RNA isolation. Primer sequences are listed in table 1.
-
Both the QB-Tn9-4s cells and its clones grew as no anchorage dependent cells. They can be maintained as a suspension culture in shake-flasks at 90 r/min or T-flasks in TNM-FH medium at 28℃. The morphology of QB-Tn9-4s generally consists of 70% spindle shaped cells with an average size of 18.2 × 57.7 μm and 30% round cells with a diameter of 22.2 μm. The QB-CL-B and QB-CL-A cells were comprised of slightly smaller spindle shaped cells which measured 18.4 × 49.4 μm to their parents (18.2 × 57.7 μm) [12]. Both the QB-Tn9-4s and two clones cells were a little bigger than Tn5B1-4 cells (16.6 × 38.4 μm) [9](Fig. 2).
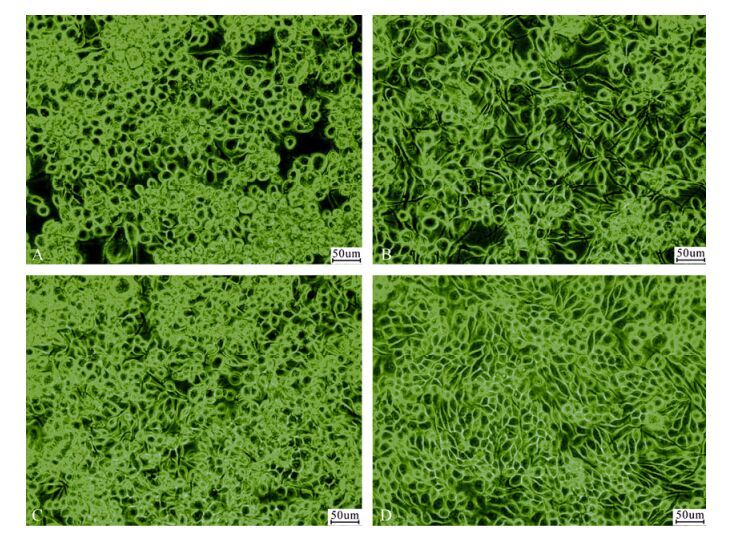
Figure 2. Phase contrast microscopic images of QB-Tn9-4s(A), QB-CL-A(B), QB-CL-B(C), and Tn5B1-4(D) cell lines. Bar = 50 μm.
-
Growth rates for the clones grown in T-flasks were determined by one-step growth curves. For these studies these cells were propagated in TNM-FH culture medium. The results indicated that QB-CL-B cells grew at a higher rate compared to the QB-Tn9-4s, QB-CL-A, and Tn5B1-4 cells, and all of the cells, QB-CL-B, QB-CL-A, Tn5B1-4 and QB-Tn9-4s, peaked at 1.65 × 106, 1.56 × 106, 1.58 × 106 and 1.49× 106 cells/mL at 5 days, respectively. Doubling times for QB-CL-B, QB-CL-A, Tn5B1-4 and QB-Tn9-4s were 20.2, 21.7, 20.9 and 22.6 h, respectively. Thus growth rates and doubling times were similar for all of these cells (Fig. 3).
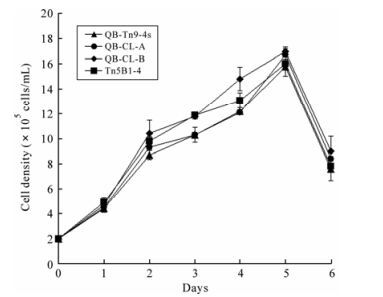
Figure 3. Growth curves of QB-Tn9-4s, QB-CL-A, QB-CL-B and Tn5B1-4 cell lines. Error bar indicate standard error of mean from three replicates.
-
The susceptibility of QB-CL-B, QB-CL-A, QB-Tn9-4s, and Tn5B1-4 cells to wild-type AcMNPV was approximately similar with infection of over 93% of the cells by the fourth day p.i.. The production of budded virus by the clone QB-CL-B appeared to be a higher titer (3.76 × 107 TCID50/mL) compared to the QB-Tn9-4s (3.37 × 107 TCID50/mL) and Tn5B1-4 cells (2.98 × 107 TCID50/mL). Similar results have been previously reported suggesting that the rate of virus titer from SF9 cells (9.24 × 107 TCID50/mL) was highest in comparison to the all of T. ni cells. The production of occlusion bodies (OBs) by T. ni cells was high, around 102--111 OBs/cell, and out-performed SF9 cell production of OBs by a factor of 2.6 (Fig. 4 and Table 2).
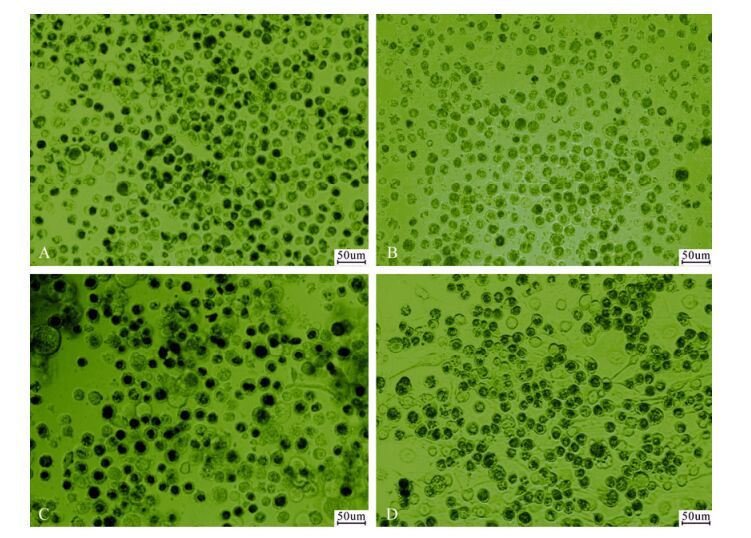
Figure 4. Phase contrast microscopic observation of infected cells from QB-Tn9-4s(A), QB-CL-A(B), QB-CL-B(C), and Tn5B1-4(D). Bar = 50 μm.

Table 2. The susceptibility of QB-CL-B QB-CL-A, QB-Tn9-4s, and Tn5B1-4 cells to Wild-type AcMNPV and viral production.
-
Production of β-galactosidase and secreted alkaline phosphatase in different cells was also compared. The clone cells, QB-CL-B, showed higher levels of recombinant protein production in comparison to Tn5B1-4, QB-Tn9-4s, and QB-CL-A cells. At seven days p.i., the production of SEAP by QB-CL-B cells out-performed Tn5B1-4, QB-Tn9-4s and QB-CL-A cells by a factor of 1.2, 3.3 and 2.1 (Fig. 5). Both QB-CL-B and QB-CL-A cells showed higher levels of β-galactosidase production in comparison to High Five and QB-Tn9-4s cells. At six days p.i., QB-CL-A and QB-CL-B cells produced 40% and 15% more β-galactosidase than High Five or QB-Tn9-4s cells, respectively (Fig. 6), but QB-CL-A cells appeared to be have a much lower level of SEAP expression.
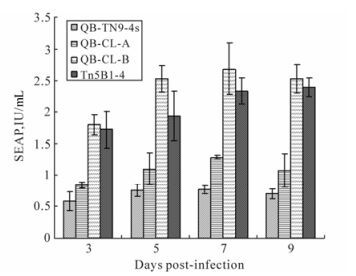
Figure 5. Secreted alkaline phosphatase (SEAP) levels were measured from QB-Tn9-4s, QB-CL-A, QB-CL-B and Tn5B1-4 cells infected with recombinant AcMNPV-SEAP virus at 3-9 days p.i.. The error bars represent standard errors of the mean from triplicate samples.
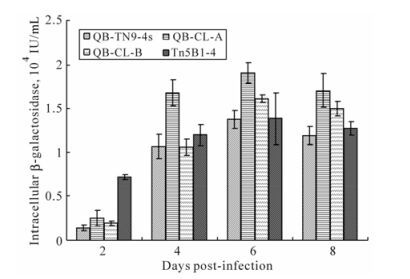
Figure 6. β-galactosidase activities in cell cultures of QB-Tn9-4s, QB-CL-A, QB-CL-B and Tn5B1-4 cell lines infected with a recombinant AcMNPV-β-gal virus. The enzyme activity in these cell cultures was determined at 2-8 days p.i.. The error bars represent standard errors of the mean from triplicate samples.
Analysis of TNCL Virus
Cell Morphology
Growth Curves of Cell Lines
Viral Susceptibility and Virus Production
Expression of Recombinant Proteins
-
In the current studies, we established a suspended cell lines from Trichoplusia ni egg tissue, QB-Tn9-4s. Some clones were selected from the new cell line and one of the clones was obtained from the parental cell line, QB-Tn9-4s at passage 91. This clone, designated as QB-CL-B, exhibited a small distinct morphology and a similar growth rate to Tn5B1-4 cells in serum-containing TNM-FH medium. Measurement of cloned cell sizes indicated that QB-CL-B cells are slightly larger than Tn5B1-4 cells but smaller than QB-Tn9-4s cells. The clone grown in TNM-FH medium is loosely adherent so it may be cultured in suspension culture or T-flasks. We also examined QB-CL-B cells in terms of growth characteristics, susceptibility to virus infection, and production of recombinant proteins. Comparisons with Tn5B1-4 and QB-Tn9-4s cells revealed that QB-CL-B cells were similar to Tn5B1-4 and QB-Tn9-4s and QB-CL-A in susceptibility to WT-AcMNPV infection (Over 93%) but the yield of BVs appeared to be different from its parental cells and Tn5B1-4 cells. The kinetics of β-galactosidase and SEAP protein production from QB-CL-B were higher than those from its parental cells and Tn5B1-4 cells after 6 days and 7 days p.i., respectively. Another cell, QB-CL-A, exhibited the highest production of β-galactosidase protein when compared to other cell lines, but expression of SEAP and rate of growth were much lower than other cells. Recently, an adventitious virus was identified in Tn5B1-4 cell lines during production of hepatitis E virus-like particles in High Five (Tn5B1-4) cells infected with a recombinant baculovirus vector [10]. We therefore examined QB-CL-B cells for the presence of the TNCL virus by RT-PCR analysis. We didn't find the TNCL virus in AcMNPV infected uncloned parental cell line (QB-Tn9-4s) or SF9 cells (a negative control), but TNCL virus was detected in AcMNPV-infected High Five cells used as a positive control. The results showed that QB-Tn9-4s cells (parental cells) were free of the alphanodavirus. So we confirmed that the new clone is free of TNCL virus and will be a valuable new tool for recombinant protein expression using the baculovirus expression system.

 DownLoad:
DownLoad: