











-
H1N1 is a highly infectious respiratory pathogen that can infect many species. It circulated in humans from 1918–1957, and then again from 1977 to the present day [1, 6], once again posing a global threat [2]. In April 2009, a pandemic H1N1 occurred initially in North America, and then spread rapidly to Europe, Asia, Australia and Africa. The World Health Organization (WHO) elevated the influenza pandemic alert level to 6, the highest alert level. As of 1 August 2010, worldwide more than 214 countries and overseas territories or communities had reported laboratory confirmed cases of pandemic influenza H1N1 2009, including 18449 over deaths (http://www.who.int/csr/don/2010_08_06/en/index.html). It was not until August 10, 2010, that the WHO Director-General Dr Margaret Chan announced that the H1N1 influenza event had moved into the post-pandemic period. In this work we analyze samples collected from this period to investigate the transmission and circulation patterns in China, with the goal of gaining insight and guidance to prepare for future influenza pandemics.
A total of 100 H1N1 real-time-PCR positive throat swabs were collected from fever patients in Zhejiang, Hubei and Guangdong provinces between June and November 2009. Virus isolation and whole-genome sequencing was performed to investigate the evolutionary relationships and mutation statuses of the pandemic H1N1 viruses. Totally, 39 HA sequences, 52NA sequences, 36 PB2 sequences, 31 PB1 sequences, 40 PA sequences, 48 NP sequences, 51 MP sequences and 36 NS sequences were obtained, including 20 whole genome sequences. All gene segments were characterized and phylogenetically analyzed based on BioEdit7.0 and MEGA version 4.0.
HTML
-
A total of 100 H1N1 flu real-time-PCR positive throat swab were collected from fever patients in Zhejiang, Hubei and Guangdong provinces between June and November 2009, by local CDC laboratories. All samples were processed and detected by Real-time-PCR for pandemic H1N1 according to the CDC protocol [3]. Next, viruses were isolated from the 57 Real-time-PCR positive samples. Madin-Darby canine kidney cells (MDCK), were cultured to a density of 75-90% in tissue culture flasks. Discard the medium (10% calf serum DMEM) and washed three times with 6 mL Hank's buffer. Swabs were washed by 200 μL 0.85% NaCl, cells were inoculated and the flasks were gently shaken. After 1~2 h incubation in 37℃ in 5% CO2 tanks, the liquids were discarded, cells were washed three times with 6 mL Hank's buffer, and then 6mL 10% calf serum DMEM with modified trypsin (TPCK 2 µg/mL concentration) medium was added. When 75%~100% cells displayed cytopathic effects, viruses were harvested by freeze and thaw. Then virus titers were tested by hemagglutination tests with 0.8% rooster erythrocyte. Negative samples were blind passaged for 2~3 generations and positive samples were stored at -80℃.
-
RNA was extracted by a Blood viral DNA/RNA Kit (Biomiga), and RT performed using a RevertAidTM First Strand cDNA Synthesis Kit (Fermentas). Reverse transcription primers were: AGCGAAAGCAGG for PB2, PB1 and PA; AGCAAAAGCAGG for HA, NA, MP, NS and NP. Primer synthesis and cDNA sequencing was performed by the AuGCT Biotechnology Company (China), (Table 1). Clustalw1.83 was used for multiple sequence alignment; Bioedit7.0 was used to edit obtained sequences; MEGA4.0 was used to draw phylogenetic tree by the neighbor joining method.

Table 1. Primer names and sequences
Sample screening and virus isolation
RNA extraction, reverse transcription and sequencing
-
A total of 39 HA sequences, 52 NA sequences, 36 PB2 sequences, 31 PB1 sequences, 40 PA sequences, 48 NP sequences, 51 MP sequences and 36 NS sequences were identified, including 20 whole genome sequences. The GenBank accession numbers are CY095718 to CY096049.
-
Following phylogenetic analysis of all eight segments, three genes were taken as representatives: HA (Fig. 1A), NA (Fig. 1B) and PB2 (Fig. 1C). The A/California/04/09 genome was taken as the reference genome and whole open reading frames (ORFs) were adopted for phylogenetic tree construction. Other representative worldwide strains were included from North America, South America, Australia, Southeast Asia, East Asia and Western Europe. All data were downloaded from NCBI. The scales denoting evolutionary distance between corresponding gene sequences were small (0.0002 or 0.0005), indicating that all the genes were highly conserved. Comparison of sequences also revealed a high degree of homology (98–99%) with the known epidemic strain A/California/04/2009 (H1N1) (data not shown). Although all sequences showed a similar level of high conservation, they clustered into distinct groups with only a few distinct strains falling outside these groups. Phylogenetic trees of PB1, PA, NP, MP and NS showed similar structure (data not shown).
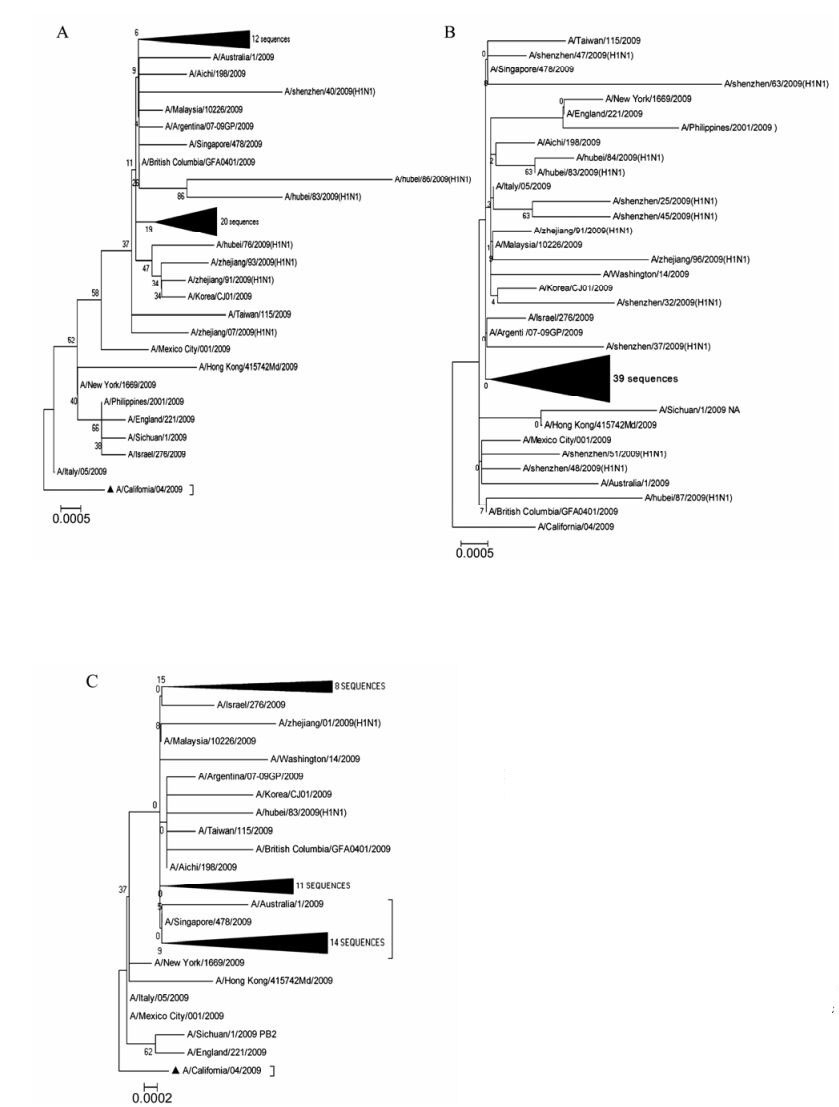
Figure 1. Phylogenetic trees of the HA (A), NA (B) and PB2 (C) Clustalw1.8 was used for multiple sequence alignment; Bioedit7.0 was used to edit obtained sequences; MEGA4.0 was used to draw phylogenetic tree by neighbor joining method.The values at the branches denoted the percentage of 1, 000 times of bootstrap re-sampling and the ratio scale denoted evolutionary distance between every two gene sequences. California/04/09 genome was taken as references and the ranges adopted for phylogenetic analysis of three genes were the ORFs.
-
The transmission ability of the influenza virus is mainly determined by the receptor binding sites (RBS), which are defined by three structural elements; a130-loop, a 190-helix site and a 220-loop [4]. Compared with A/California/04/2009, the virus isolates were conserved at the 130-loop and 190-helix sites: VTAA (135-138) at loop 130 and DQQSLYQNA (190-198) at helix 190. However, at the loop 220(221-228), there were 3 mutations in the HA sequences (Fig. 2): A/Hubei/ 86/2009PKVRDQEG→PKVRDQEA, A/Zhejiang/08/ 2009 PKVRDQEG→PKVRDQER, A/Hubei/75/2009 PKVRDQEG→PKVRDQGG. The A/Hubei/75/2009 was isolated from an acute case, while the other two were from patients with mild symptoms.

Figure 2. Key sites of HA, nucleotide sequences were edited and then translated by Bioedit7.0, and the graph was processed by CorelDRAW12.
-
Amino acids 119, 274, 292 and 294 amino acids of NA protein are associated with resistance to neuramidinase inhibitors, either a 274(H) or 294(N) mutation would confer resistance [11, 12, 15, 16]. All the newly sequenced viruses were conserved at all these positions (Fig. 3A), indicating sensitivity to neura midinase inhibitors.

Figure 3. Key sites of NA M2 and PB2: A was for NA, B was for PB2, C was for M2. Nucleotide sequences were edited and then translated by Bioedit7.0, and the graph was processed by CorelDRAW12.
Amino acid 627 of the PB2 protein is associated with virulence to mammalians [5, 13]. 627 Glu is a distinguishing characteristic of avian viruses, while the E→ K mutation can enhance virulence to mammals. All the isolates had Glu at 627 (Fig. 3B), consistent with the avian virus origin.
M2, the trans-membrane protein, acts as an ion tunnel. The amino acids at sites 26, 27, 30, 31, 34 have been associated with resistance to adamantanes [7, 9, 14]. All isolates reported in this study possessed the mutation S→N (Fig. 3C), indicating resistance to adamantanes.
-
All the isolates, as well as all other sequenced strains, except for A/California/04/09, had the P88S mutation in the HA gene (Fig. 4A). A similar mutation was also found at P224S of PA (data not shown).

Figure 4. Other key site amino acid mutations. HA P88S mutation (A); NA V106I mutation (B); NP V100I mutation (C); T202A and S208T HA mutations (D). A/California/04/09 was the reference and seven representative China isolates were added to the database. Nucleotide sequences were edited and then translated by Bioedit7.0, and graph was processed by CorelDRAW12.
All the 52 NA sequences contained the V106I mutation of the NA gene (Fig. 4B). Of the total 86 sequences in the database, 12 were conserved, all of which were among the earliest isolates, including the first China isolate A/Sichuan/1/09 (2009-5-10). The V106I mutation found in all the isolates corresponded to that seen in one of the early isolates A/Mexico City/001/09 (isolated 2009-5-7). Similar mutations were also detected at N248D of NA and I123V of NS.
Six out of the total of 48 NP sequences were conserved in terms of V100I mutations (Fig. 4C), including strains isolated from both the first (such as A/Sichuan/1/09) and second wave (such as A/Zhejiang/98/09, November 2009) of the 2009 pandemic in China.
All our HA sequences contained the T202A and S208T mutations (Fig. 4D). It was notable that of the total 73 sequences in the database, 15 had the T202A mutation with a conserved 208 site, while two (A/Taiwan/115/09 and A/Karaj/6072/10) had the S208T mutation with a conserved 202 site.
Other sporadic mutations that appeared 5 times or more were shown in Table 2. What most deserved the attention were the mutations that occurred at sites 75-78 LSTA→FSTS; the L75S and the A78S always occurred in tandem, suggesting co-variance.

Table 2. Other sporadic mutations that appeared 5 times or more
Besides, we isolated one strain (A/zhejiang/93/2009) that was conserved at 321 of HA (Fig. 5), while others possessed the mutation I321V. It was collected at October 12, 2009, thus belonged to the second pandemic wave.

Figure 5. HA I321V mutation. Nucleotide sequences were edited and then translated by Bioedit7.0, and graph was processed by CorelDRAW12.
Virus isolation and sequencing
Phylogenetic tree of the genomes
Key sites of HA
Key sites of NA, PB2 and M2
Other amino acid mutations
-
Influenza viruses possess a segmented, negative single-stranded RNA genome. The low-fidelity RNA polymerase results in a high replication-error rate, which, together with reassortment and recombination, contributes to the high genetic diversity among influenza viruses [8]. These genetic shifts lead to the rapid emergence of novel antigenic strains. It is therefore vital to maintain continuous surveillance of the presence of H1N1 stains.
As part of a H1N1 surveillance program in China, throat swabs were collected from patients between June and November 2009 and isolated viruses were sequenced. Phylogenetic analysis of the eight segments showed that all they shared close homology with strain A/California/04/2009. But the sequences clustered into groups with few exceptions. These clustered strains dominated in the three investigated regions, indicating they possess certain fitness advantages over other variants.
Key sites analysis showed that 130-loop, 190-helix of HA; 119, 274, 292 and 294 amino acids of NA and 627 of PB2 protein were conserved in almost all the sequences, and the M2 protein had the S32N mutation. But, at the loop 220(221-228) of the HA receptor binding sites, there were 3 substitution in 39 total HA sequences. Of these, one was isolated from an acute case, while the other two were from patients with mild symptoms, so the significance of these changes is unclear. However, the A/Hubei/75/2009 E227G mutation may influence the binding specificity since it was located at the RBS [10].
Despite numerous studies of H1N1 evolution, the transmission and circulating pattern of the influenza virus haven't fully understood. In this paper, we analyzed the iconic mutations. In the HA P88S mutation (Fig. 4A), all the isolates along with all the other strains around the world mutated except the A/California/04/2009, a similar mutation was also found in P224 S of PA. This suggests that these mutations were advantageous, or at least neutral, and that they arose at the very early stage of the pandemic, before its introduction into China, after which they spread rapidly.
We suspect that the China H1N1 isolates might have had multiple origins, in the NA V106I mutation (Fig. 4B), although all the 52 sequences mutated compared with the A/Califronia/04/09, we noticed that in the total 86 sequences in our dataset, 12 were conserved, all of which were early isolates including the first China isolates A/Sichuan/1/09(2009-5-10). The V106I mutation in the isolates matched that in one of the early isolates, A/Mexico City/001/09 (isolated on 2009-5-7). It was believed that the pandemic H1N1 originated from North America, and this could thus provide evidence of multiple introductions into China.
In the circulating of the virus, the predominance of a particular sub-strain was progressive. The NP V100I mutations also support this hypothesis: six of the 48 sequences were conserved, and the conserved sequences appeared in isolates from both the first (such as A/Sichuan/1/09) and second waves (such as A/Zhejiang/98/09, November 2009) in China.
It was interesting that in the 73 total HA sequences data base we set, 15 had the T202A mutation with a conserved 208 site, and two (A/Taiwan/115/09 and A/Karaj/6072/10) had the S208T mutation with a conserved 202 site, while all the others were mutated at both sites. This might be the result of recombination by co-infection in certain cases, or simply be the result of random mutations. It was notable that these sites were between the 190 helix site and the 220 loop of the HA RBS, and we could therefore not exclude the possibility that the evolution of this sequence had been affected by extensive vaccine use. The high conservation of the pandemic H1N1 and the lack of associated clinical data indicated the need for further studies.
The HA I321V mutation, which had a similar distribution to the HA P88S mutation, was not found in one strain (A/Zhejiang/93/2009) on October 12, 2009. However, all the other sites (NP (V100I), NA (V106I, N248D) NS (I123V)) were mutated, suggesting that this might represent a reverse mutation.
Although the H1N1 pandemic has passed, the influenza virus threat remains. The isolation and analysis could provide significant information about the transmission and circulation patterns of this virus. This could in turn provide helpful insight into suitable measures for the preparation and control of subsequent pandemics.

 DownLoad:
DownLoad: