











-
Hand, foot, and mouth disease (HFMD) is a common illness in children. However, no effective vaccine or antiviral drug is yet available for HFMD. Coxsackievirus A16 (CVA16) and enterovirus 71(EV71) are two major etiological agents of HFMD [1, 10, 28]. The seroprevalence for individuals (ages≥1 year) was found to be nearly 60% for CVA16 and 40% for EV71 [26]. In recent years, large outbreaks of EV71-associated HFMD in the Asia-Pacific region [3, 13, 25], often coupled with severe clinical manifestations, have drawn a lot of attention to this virus. In contrast, CVA16 appears to have drawn very little interest, probably due to its association with often mild and benign clinical symptoms. As such, very little information has been made available for CVA16. However, it has been observed the recombination between CVA16 and EV71 [32, 34], and the co-circulation of these two viruses may have contributed to the increase of HFMD cases in China over the past few years [33]. Thus, for CVA16, further understanding of its virology, epidemiology and virus-host interactions and host pathogenesis is of importance.
CVA16 is described as one of the human enterovirus A species under the genus Enterovirus in the Picornaviridae family of viruses, which includes EV71 and poliovirus [20]. The exact mechanism for CVA16 replication has been poorly understood and was often speculated based on studies on other picornaviruses. CVA16 virus particle first attaches to the host cell surface via cellular receptors (HSCARB2 and PSGL-1) [23, 29]before entering and uncoating to unveil the viral RNA genome. Viral RNA is translated by cellular translational machinery to give a polyprotein that is then cleaved by the virus-encoded proteases 2Apro and 3Cpro to give four structural (VP1-4) and seven non-structural (2A-C and 3A-D) individual proteins [18]. Within virus-induced membrane vesicles, viral RNA (+) is copied by the viral RNA polymerase, 3Dpol, to give (-) strand RNA intermediates, which in turn provide the template for the synthesis of (+) strand viral RNA. The (+) strand viral RNAs are used to generate more (-) strand viral RNAs, which are translated into viral proteins or packaged into progeny virions. Lysis of host cells will result in the release of progeny virions into the cytoplasm, where it directly translates into a polyprotein [35].
Viral kinetic studies have elucidated the process of host-viral interactions in influenza A, hepatitis B and C, and cytomegalovirus infections [2, 8, 11, 21, 27]. These detailed viral studies have enabled investigators to examine the efficacy of antiviral compounds in limiting viral replication while assessing the development of resistance with resurgence in viral replication. In the Picornavirus family, the kinetics of poliovirus [6], Enterovirus 71 [15] and Hepatitis A Virus (HAV) [5] have been described in several studies. However, little information is known about CVA16 infection. The objectives of the present investigation were to conduct a study on viral RNA replication, viral protein synthesis, packaging and secretion in rhabdomyosarcoma (RD) cells to achieve an understanding of the viral kinetics of CVA16.
HTML
-
RD cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum plus 100 IU of penicillin, and 100 μg of streptomycin per mL. CVA16 strain HN1903/CHN/2010 (B1b genotype) was obtained from Henan Provincial Center for Disease Control and Prevention, Zhengzhou, China. To prepare virus stocks, viruses were propagated on a confluent RD cell monolayer in DMEM with 2% FBS. Virus titers were determined by plaque assay in RD cells as described previously [12].
-
Confluent RD cells were infected with nil or CVA16 at MOI 0.1 and 1 for 1 h at 37℃, respectively, after which infected cells were washed twice with phosphate buffered saline (PBS) to remove excess virus and cultured in fresh DMEM medium containing 10% FBS until used in subsequent experiments.
-
The cell morphology was monitored and recorded using a phase-contrast microscope (Evosxl, AMG) at different time points [30]. The cell viability upon CVA16 infection was determined by MTT assay as described elsewhere [14]. The optical density (OD) of the wells was measured using a microplate spectrophotometer (iMarkTM Microplate Reader, BIO-RAD) at 490 nm.
-
RNA extraction from infected cells and culture supernatants was performed by TRIzol reagent (Invitrogen, USA) and chloroform, and precipitated by isopropranol. Purified RNA was resuspended in distilled water [22]. The intracellular virus RNA was extracted as described elsewhere [15]. To set up the standard curve of infectious viruses, the CVA16 viral particles were first purified. Briefly, the infected cells were lysed by two rounds of freezing and thawing. The resultant lysates were filtered through a 0.22-μm membrane filter. CVA16 viral particles were then purified from the resultant suspension by centrifugation at 28, 000 rpm for 2.5 h at 4℃ in a Beckman SW40 rotor with 1 mL of 30% sucrose cushion. Titers were then determined by plaque assay in RD cells. Then the viral RNA was extracted from those purified CVA16 virus stocks. RNA was diluted at ten-fold serial in distilled water and used to reflect the calculated PFU from 10 to 1 ×105 live virions.
-
Reverse transcription was carried out in a 20 μL volume containing 5 μL of RNA extracted from samples or from ten-fold serial diluted virus RNA standard (from 10 to 105 copies) using a PrimeScript® RT Kit (Takara, Japan) according to the manufacturer's instructions. The quantitative real-time polymerase chain reaction (qPCR) was carried out using the Chromo4 Real-Time PCR system (BIO-RAD) with RealMaster Mix(SYBR Green) Kit (Takara, Japan) and specific forward CVA16-5'NTR-F (5'-TCCTCCGGCCCCTGA-3') and reverse CVA16-5'NTR-R(5'-AATTGTCACCATAAGCAGCCA')primers targeting a conserved region of the 5'NTR [22]. qPCR assay was carried out in a 20 μL volume consisting of 9 μL of 2.5× RealMaster Mix/20×SYBR Green solution containing HotmasterTaq DNA polymerase, 1 μL of 5 μmol/L of each oligonucleotide primer, and 4 μL of cDNA template. The target fragment amplification was carried out as follows: initial activation of Hotmaster Taq DNA Polymerase at 95℃ for 2 min; 45 cycles in four steps: 95 ℃ for 15 s, 57 ℃ for 15 s, and 68 ℃ for 20 s.
-
Western blot was performed as described elsewhere [17]. Briefly, cell lysates were mixed with SDS-PAGE sample buffer, boiled, and separated on 13.5% polyacrylamide gels. Proteins were transferred onto PVDF membranes for Western blot analysis. Membranes were probed with mouse anti-VP1 polyclonal antibody (a gift kindly provided by National Institutes for Food and Drug Prevention and Control, China) or mouse anti-tubulin purified antibody (Covance, USA) diluted 1:1000 in PBST plus 1% skimmed milk followed by a corresponding AP-conjugated secondary antibody (Sigma, St. Louis, MO, USA) diluted 1:1000. Positive signals on the membranes were developed by chemiluminescence using NBT (Dinguo, China) and BCIP (Dinguo, China).
-
The results are from at least three replicates and are reported as mean ± SD. All statistical analyses were carried out with the GraphPad Prism software package. Groups were compared by using the Student's t-test. The p value < 0.05 was taken to indicate statistical significance.
Cells and viruses
Virus infection
CPE analysis and Cell viability assay
Extraction of CVA16 RNA
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Western blotting
Statistical analysis
-
To investigate the effects of CVA16 infection upon host cells, RD cells were subjected to infection by CVA16 at either a MOI 0.1 or 1, followed by observation under a phase-contrast microscope. After infection for 12 h, the infected cells started to exhibit a visible cytopathic effect reaction with morphological changes, including rounding up, shrinkage, and detachment from the culture medium (Fig. 1A, Ⅶ and Ⅺ). Later on, the cells infected with CVA16 both at MOI 0.1 and 1 underwent rapid cell death and detached from the culture medium (Fig. 1A, Ⅷ and Ⅻ). More cells exhibited CPE when infected at MOI 1 compared to the cells infected at MOI 0.1 at 12 h and 24 h p.i. (Fig. 1A, Ⅶ, Ⅺ, Ⅷ and Ⅻ), respectively. CPE was not observed in control cultures which were mock treated at each indicated time point (Fig. 1A, Ⅰ, Ⅱ, Ⅲ and Ⅳ).
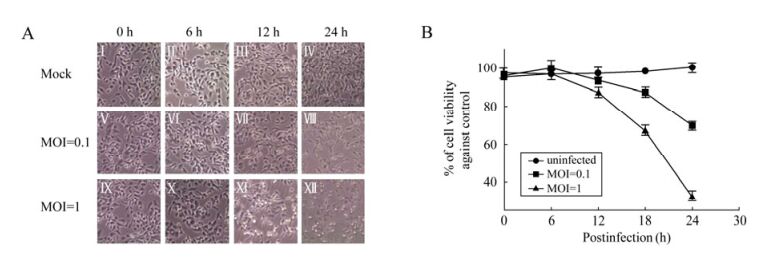
Figure 1. CPE analysis and Cell viability assay upon CVA16 infection. RD cells were infected with CVA16 at MOI=0.1 or MOI=1. A: The cell morphology was monitored and recorded using a phase-contrast microscope at 0, 6, 12, 24 h p.i. (original magnification, × 100); B: Cell viability was measured by MTT assay at 0, 6, 12, 18, 24 h p.i. Each point represents the mean ± SD of at least three separate experiments performed in duplicate.
Cell viability at each time point was quantitatively confirmed by performing the MTT assay. As shown in Fig. 1B, the cells infected with CVA16 at MOI 0.1 or 1 did not significantly decrease at 12 h p.i.. However, at 18 h p.i., the viability of the cells in the MOI 1 group rapidly decreased by almost 35% compared with the control group and only 15% in the MOI 0.1 group. At 24 h p.i., the percentage of viable cells was significantly reduced to almost 30% compared with the control group in the MOI 1 group and to around 70% in the MOI 0.1 group.
-
To establish the growth curve for CVA16 in RD cells, the cells were infected with infectious CVA16 at either MOI 0.1 or 1. Virus adsorption was performed at 37℃ for 1 h to permit virus binding and entry into host cells. At various times p.i., the total intracellular viral RNA (Fig. 2B), the intracellular viral RNA that was packaged (Fig. 2C) into virus particles, and the extracellular viral RNA (Fig. 2D) were determined by qRT-PCR. Virus antigen expression was monitored by Western blotting analysis of VP1 proteins (Fig. 2A).
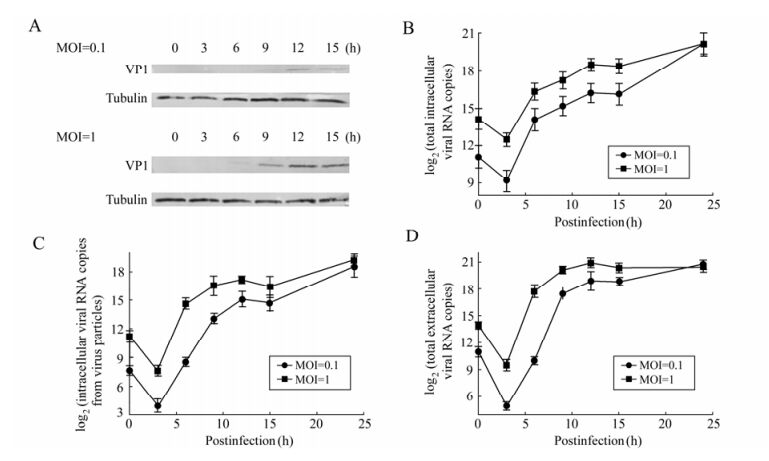
Figure 2. Replication kinetics of CVA16 in RD cells. A: Western blotting analysis of VP1 at different time points p.i. Total protein extracts of RD cells infected with CVA16 at MOI 0.1 or MOI 1 were tested against the mouse anti-VP1 antibodies. The Tubulin housekeeping protein was included to ensure equal loading of protein samples. B: Time course of total intracellular viral RNA synthesis from RD cells. RD cells (1×104) were infected with CVA16 at MOI 0.1 or MOI 1 for 1 h. Total intracellular viral RNA copy was determined by qRT-PCR at 3, 6, 9, 12, 15, 24 h p.i. C and D, Time course of infectious CVA16 production from RD cells. RD cells (1×104) were infected with CVA16 at MOI 0.1 or MOI 1 for 1 h. Virus RNA copy in both cell lysates (intracellular) (C) and supernatants (extracellular) (D) were determined by qRT-PCR at 3, 6, 9, 12, 15, 24 h p.i. CVA16 RNA copy from uninfected cells was zero at all indicated time points. Each point represents the mean ± SD of at least three separate experiments performed in duplicate.
In order to examine the kinetics of the total intracellular viral RNA synthesis, we employed qRT-PCR to quantitatively determine viral RNA copy number at each time point. The results of qRT-PCR are shown in Fig. 2B, and indicate that little viral RNA was detected between 0 to 3 h in cells infected with CVA16 both at MOI 0.1 and 1; thereafter the amounts of total intracellular viral RNA of the two groups were constitutively increased between 3 and 12 h. The exponential phase was from 3 to 6 h p.i. and then remained almost unchanged at 15 h p.i. in both infected groups. The total intracellular viral RNA maintained the same levels at 24 h p.i. in both infected groups.
CVA16 infected cells were harvested at each time point to perform Western blotting. The results (Fig. 2A) showed that the level of viral VP1 proteins failed to reach detectable levels in infected cells until 6 h p.i. at MOI 1 and 9 h p.i. at MOI 0.1. Thereafter the VP1 levels showed a rapid increase and then reached a peak at 12 h p.i. in both groups. Further time points were not examined. Obviously, the VP1 levels were much higher in the cells infected at MOI 1 than at MOI 0.1. In the case of infection at MOI 1, the VP1 level was maintained until 15 h p.i.; whereas the VP1 protein level was decreased after 12 h p.i. when the cells were infected with CVA16 at MOI 0.1.
To examine the kinetics of viral packaging, we first isolated intracellular virus particles from host cells at each time point to measure the virion RNA by qRT-PCR. The results (Fig. 2C) showed that the intracellular virions underwent a significant decrease for the first 3 h p.i. Thereafter, the intracellular virions began to increase and enter into the exponential phase until 6 h p.i. at MOI 1 and until 9 h p.i. at MOI 0.1. The viral RNA package peaked at 12h p.i. for both the MOI 0.1 and MOI 1 infected groups. The total amounts of intracellular virions then decreased gradually from 12 to 15 h p.i. The intracellular CVA16 virions maintained the same levels at 24 h p.i. in both infected groups.
To examine the kinetics of viral secretion, we first isolated extracellular virus particles from culture medium at each time point to measure the virion RNA by qRT-PCR. The results (Fig. 2D) showed that little extracellular virus was detected between 0 to 3 h in cells infected with CVA16 both at MOI 0.1 and 1; thereafter the amounts of extracellular virus in the cultures of the two groups increased between 3 and 12 h. However, the virus secretion entered into the exponential phase from 3 to 9 h p.i. at MOI 0.1 while the exponential phase was only lasted 3 h from 3 to 6 h p.i.at MOI 1. In both cases, the secreted virions peaked at 12 h p.i. in the culture medium. Thereafter, the virion secretion declined and even stopped. For cells infected at MOI 0.1 or 1, the virions in the culture media were similar at 24 h p.i.
CPE analysis and Cell viability assay
Growth Curve of CVA16 in RD cells
-
Viral kinetic studies have provided a deeper understanding of the natural history of viral infections in humans with establishment of latent reservoirs, viral shedding, and viral clearance [4, 24]. In addition, studies on viral load have shed light on the probability of viral transmission from mother to infant [7, 31] and sexual transmission from one person to another [9]. More recently, these viral kinetic studies have stimulated a newly emerging field regarding the effectiveness of antiviral agents on viral replication as a surrogate in modeling their population transmission effects. Although these viral kinetics were examined in vivo, investigation of CVA16 kinetics in vitro at the cell level is also of importance, since the virology, epidemiology, virus-host interactions and host pathogenesis of CVA16 are poorly understood. In this study, the kinetics of viral replication, viral protein expression, package and secretion as well as the effects of viral activities at the cell level were carefully examined at different time points, to set up a solid base for investigating the viral kinetic of CVA16 in vivo that could help further study virus-host interactions and host pathogenesis of CVA16 in the future.
Utilization of qRT-PCR have enabled investigators to accurately measure the quantitative amount of viral nucleic acids produced within a host over time, thereby enabling investigations into virus-host interactions and host pathogenesis [16, 19]. In this study, we also employed qRT-PCR, which serves as a rapid and efficient molecular biology technology to quantitatively determine DNA copy number with high sensitivity and specificity, to determine viral RNA copy number at each time point p.i. To perform qRT-PCR, we first quantified the inoculated virus by plaque assay in RD cells to set up the standard curve of infectious viruses. Thereafter, standard and sample virus cDNA synthesized from RNA preparation with reverse transcriptase were serially diluted and then subjected to measurement with qRT-PCR. The exact viral RNA copy number at each time point was finally calculated by comparison with a standard curve prepared from cDNA solutions corresponding to the serially diluted solutions of standard viral RNA, and the growth curve for CVA16 in RD cell was then established by using these viral RNA copy numbers. Therefore, qRT-PCR assay can enable viral kinetic studies of CVA16 and other picornaviruses.
As shown in Fig 2B and 2D, both the total intracellular RNA copy number and extracellular RNA copy number at 0 h p.i. were estimated to be 1×104 copies per well at MOI 0.1 and 1×105 copies per well at MOI 1. The results suggested almost 50% virions entered into host cells after virus adsorption at 37℃ for 1 h. At 3 h p.i., the total intracellular RNA copy number was significantly decreased by almost 75%, suggesting that most of the viral RNA was degraded immediately after its entry into host cells by RNases, and the virus replication was hindered by host cells at 3 h p.i. Therefore, it is speculated that CVA16 would first attach, uncoat and enter into the host cell via its specific receptors after adsorption for 1 h. Later on, the amounts of total intracellular viral RNA, intracellular viral RNA that was packaged into virus particles, and extracellular viral RNA of the two groups were constitutively increased between 3 and 12 h (Fig. 2B, 2C and 2D). For the total intracellular viral RNA, the exponential phase was from 3 to 6 h p.i. in both infected groups (Fig. 2B). In the meantime, the viral protein synthesis was started along with the viral RNA replication, as VP1 proteins were clearly detected by Western blotting at 6 h in cells infected at MOI 1 (Fig. 2A). VP1 proteins were not detected at 6 h in cells infected at low MOI of CVA16 (MOI 0.1) (Fig. 2A). The VP1 levels were much higher in the cells infected at MOI 1 than at MOI 0.1. This suggested that more viral proteins were produced in cells infected with higher MOI. Due to the fast viral RNA replication and protein synthesis from 3 to 6 h p.i., virus package and secretion was also initiated during this phrase. As shown in Fig. 2C and 2D, a significant increase of virus package and secretion was observed from 3 to 6 h at MOI 1 and to 9 h at MOI 0.1. At 6 h p.i., about 1.5% (MOI 0.1) to 20% (MOI 1) of viral RNA was packaged into virus particles (Fig. 2C and 2D). Although the virus was rapidly replicating and secreting, the host cells were generally viable during this period. From 6 to 12 h p.i., some cells exhibited CPE (Fig. 1A), and the viral replication entered a static stage as the total intracellular viral RNA only increased by about 4-fold at MOI 0.1 and MOI 1(Fig. 2B).
Comparison of the kinetics of intracellular and extracellular virus loads, we observed different behaviors of infection at MOI 1 and MOI 0.1. At MOI 0.1, only 10% of cells were internally infected while over 90% cells were infected at MOI 1. At 6 h p.i., the ratio of the intracellular viral load of MOI 1 to MOI 0.1 was about 128 fold; however, the ratio of extracellular viral loads of MOI 1 to MOI 0.1 was about 1024 fold. At 12 h p.i., the ratio of both intracellular and extracellular viral loads of MOI 1 to MOI 0.1 was reduced to about 10 fold. These results suggest that the secreted virions from 3 h to 6 h reentered the host cells and conducted the second round infection at MOI 0.1, while the second round infection was not significant at MOI 1 (Fig. 2C and 2D). Following viral protein synthesis, the intracellular virions rapidly increased and entered into the exponential phase until 6 h p.i. at MOI 1 and until 9 h p.i. at MOI 0.1 (due to the second round infection), respectively. In both cases, the intracellular virions reached a peak and about 40% of viral RNA was packaged into the virions at 12 h p.i. (Fig. 2C vs Fig. 2B). At MOI 0.1, the virions secreted in the supernatants increased about 24-and 100-fold from 3 to 6 h, and 6 to 12 h, respectively; while they increased 128-and 8-fold during the same periods at MOI 1. Thereafter, the virion secretion slightly increased from 12 h p.i. to the end of the observation period at MOI 0.1 while virion secretion declined after 12 h p.i. and the total virions in the supernatants were unchanged from 12 to 24 h p.i in the cells infected at MOI 1. We showed here that the intracellular virions continued to increase after 15 h p.i. In the mean time, the ratio of packaged viral RNA to total viral RNA was constant at around 40% at both MOI 0.1 and MOI 1.
In summary, we have established a viral kinetics model of CVA16 in human RD cells (Fig. 3). CVA16 is speculated to first attach, uncoat and enter into the host cell via its specific receptors after adsorption for 1 h. Later on, CVA16 undergoes rapid replication from 3 to 6 h at MOI 1 and until 9 h at MOI 0.1. At MOI 0.1, CVA16 initiated the secondary infection as the virions were secreted before 9 h p.i. and the structure protein level decreased after 12 h p.i. Thus, our study provides important information for further investigation of CVA16 in order to understand better and ultimately control infections with this virus.
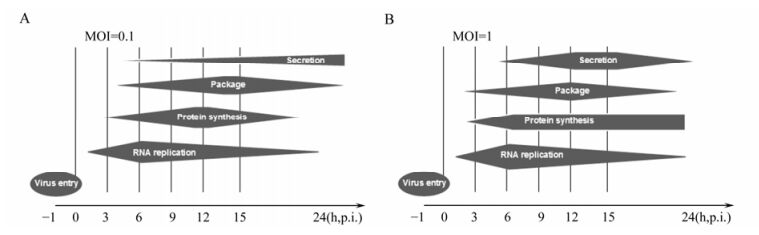
Figure 3. Scheme of CVA16 activity in RD cells at a MOI 0.1(A) or MOI 1(B).

 DownLoad:
DownLoad: