









HTML
-
Ebola and Marburg viruses are negative sense ssRNA viruses of the family Filoviridae (Feldmann H, et al., 1993). Natural filovirus outbreaks occur in parts of the Philippines and central Africa, with the most recent being in Uganda and the Democratic Republic of Congo in 2011 and 2012 (World Health Organization; 2012a and 2012b). Infection carries with it a mortality rate of up to 90%, and there is currently no approved vaccine or therapeutic for infection (Warfield K L, et al., 2009). Several vector-based vaccines are currently under investigation, including an adenoviral vector and a replication incompetent Venezuelan equine encephalitis replicon system (Reviewed in (Bradfute S B, et al., 2011)). These have shown promise in nonhuman primate (NHP) challenge models and elicit a strong immune response, but safety concerns may complicate their further development. The virus-like particle (VLP) vaccine platform shows a great deal of promise for the development of a safe and effective filovirus vaccine. It is highly efficacious in the mouse, guinea pig, and NHP models of viral infection.
-
The filovirus VLP vaccine has been produced in a mammalian cell line and in an insect cell line, the latter approach utilizing a baculovirus vector for expression of viral proteins (Warfield K L, et al., 2007). Baculoderived VLP were shown to be efficacious in both small animal and NHP models of infection and were pursued for a time due to ease of production and high immunogenicity. Potential regulatory constraints of baculo-derived products, as well as the unknown influence of insect antigen that could potentially be included in the particles, however, has resulted in a return to a human cell-line based production system. Production in human cell lines is thought to provide a more authentic mimic of live filoviruses than does production in the baculovirus system. The processing and glycosylation of the viral protein components that would occur in natural infection are better preserved in mammalian-derived VLP than in baculo-derived VLP, as is visible by western blot (Warfield K L, et al., 2007).
VLP are currently produced by transfection of 293 cells with plasmids encoding the viral matrix protein, VP40, and glycoprotein (GP) (Bavari S, et al., 2002; Swenson D L, et al., 2004; Warfield K L, et al., 2003). Plasmid sequences are derived from the Ebola Zaire 1995 strain (Zaire VLP, eVLP), Sudan Boniface Ebola (Sudan VLP), or Marburg Musoke (Marburg VLP, mVLP) viruses, but can be easily interchanged during production. Electron microscopic analysis of VLP after anti-GP immunogold staining reveals high levels of GP along the length of VLP particles, which closely resemble Ebola and Marburg viral particles (Fig. 1). The particles are approximately 1-2 µm in length and have a diameter of 50-100 nm, comparable to live viral particles (Swenson D L, et al., 2004).
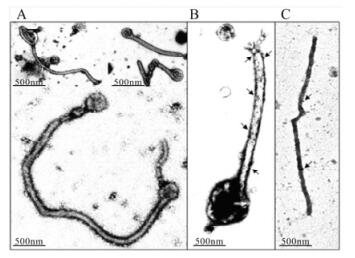
Figure 1. Electron microscopic analysis of VLP containing MARV GP and VP40. Particles obtained by ultracentrifugation of the supernatants of 293T cells transfected with both Marburg GP and VP40 were negatively stained with uranylacetate to reveal the ultrastructure (A), or labeled with anti-MARV GP monoclonal antibody (B), or anti-MARV VP40 monoclonal antibody followed by immunogold rabbit anti-mouse Ab (C) (Swenson D L, et al., 2004).
VLP will form upon VP40 transfection alone, but inclusion of GP enhances VLP egress, and GP appears to be the primary immunogenic component of the VLP (Licata J M, et al., 2004). In a study using heterologous VLP expressing Marburg GP and Ebola VP40 (m/e-VLP) or the reverse (e/m-VLP), guinea pigs were vaccinated and then challenged with either Ebola or Marburg virus (Swenson D L, et al., 2005). Animals were protected from challenge by VLP expressing the viral GP of the challenge material, regardless of the origin of the VP40. Additionally, the efficacy of VLP, as defined by the protection of mice from lethal challenge, correlates with GP content, not total protein. This is apparent from the results of vaccination studies using lots of VLP with differing GP concentrations (unpublished data). Due to the importance of GP, the GP content of VLP is quantified by western blot and ELISA analyses, and vaccinations are administered based on GP protein concentration.
-
VLP show considerable immunogenicity in the mouse and guinea pig, eliciting high anti-GP antibody titers and protecting animals from otherwise lethal challenge with mouse-adapted (ma-) and guinea pig-adapted (gpa-) Ebola Zaire (EBOV) and Marburg (MARV) viruses (Warfield K L, et al., 2004; Warfield K L, et al., 2005). Without adjuvant, intramuscular (IM) vaccination with Ebola Zaire VLP consistently confers 100% protection in C57BL/6 mice; administering a vaccine boost three weeks after the initial vaccination greatly increases antibody titers and results in protection at lower dose levels (unpublished data).
In addition to serving as an effective vaccine that confers protection via antigen-specific adaptive immune responses, VLP administered immediately prior to or after viral challenge can confer protection via innate adaptive immunity. Natural killer (NK) cells appear to play a critical role in this protection. Unlike wild type C57BL/6 mice, NK cell-deficient mice were not protected from Ebola virus challenge when VLP were administered three days prior to challenge, and transfer of VLP-stimulated NK cells protected animals from inoculation with virus (Warfield K L, et al., 2004).
One aim in the development of filovirus vaccines is the development of a pan-filovirus vaccine that will be effective against multiple filoviruses, including Marburg virus and the various Ebola viruses. VLP mixing studies have indicated that immune responses to the Marburg and Ebola GP do not interfere with each other. Vaccination of guinea pigs with a mixture of Ebola and Marburg VLP resulted in high antibody titers to both antigens and survival after challenge (Swenson D L, et al., 2005). This research paves the way for the possibility of multivalent filovirus vaccine development.
-
VLP, in combination with adjuvant, have been shown to protect NHP from lethal challenge with Ebola and Marburg viruses. Vaccination strategies have included vaccination with PolyICLC (Hiltonol), QS21, and RIBI as adjuvants, and typically utilize a vaccination with two or three boosts, spaced four to six weeks apart. A lethal challenge is generally administered as 1, 000 pfu; though the LD50 in this model has not been defined, data from the mouse model of infection indicates that the LD50 of filoviruses may be as low as 1 pfu (Bray M, et al., 1998). In recent studies, vaccination and challenge are typically administered intramuscularly (IM); IM administration of baculo-derived VLP has also provided protection in cynomolgus macaques upon aerosol challenge (unpublished work).
In one Ebola challenge study, cynomolgus macaques were vaccinated three times, IM, with 250 µg of eVLP (total protein) and 0.5 mL of the adjuvant RIBI. Antibody titers plateaued after the second vaccination and antibody was shown to be multifunctional with neutralization, antibody-dependent cytotoxicity, and complementdependent cytotoxicity capabilities (Warfield K L, et al., 2007). Animals were then challenged with 1, 000 pfu of Ebola Zaire virus. With the exception of a slight temperature elevation in two of the five animals (2-3 ºF) after challenge, the animals exhibited no symptoms or changes in temperature, and virus was never isolated after challenge (Warfield K L, et al., 2007).
mVLP have been shown to protect against a heterologous group of Marburg viruses in NHP. Guinea pigs were protected from challenge with three different Marburg viruses after vaccination with mVLP, and this study was then conducted using macaques. Nine cynomolgus macaques were vaccinated with Musoke-based Marburg VLP in combination with the adjuvant RIBI. Animals received three vaccinations, forty-two days apart, of 1 mg of VLP (total protein) and 0.1 mL of QS-21. Twenty-eight days after the final vaccination, vaccinated animals were challenged with 1, 000 pfu of one of three Marburg viruses via the subcutaneous route, with three vaccinated animals in each challenge group. The challenge viruses were Ci67, Musoke, and Ravn, and these viruses exhibit greater than 20% genetic diversity in GP (Swenson D L, et al., 2008). All vaccinated macaques achieved high antibody titers against the three viruses and survived challenge.
More recent work aims to identify the 50% effective dose (ED50) of VLP in NHP. A study utilizing three vaccinations with 100 µg of PolyICLC as adjuvant have demonstrated 100% protection from challenge with Marburg virus at low VLP dose levels. IgG antibody titers against GP were high in vaccinated animals, ranging from 4 to 6 logs, and effectively plateaued after the second vaccination (Fig. 2). Work is currently underway to explore efficacy at lower dose levels of both VLP and adjuvant.

Figure 2. Anti-glycoprotein IgG antibody titers. Eighteen cynomolgus macaques were vaccinated IM with either saline (n=3) or Marburg VLP at three different dose levels (n=5 per group). Dose levels were 10 µg, 55 µg, or 269 µg of VLP based on GP content, and animals were also administered 100 µg of PolyICLC with the vaccinations. Vaccinations took place on days 0, 28, and 56 (indicated by arrows), and animals were challenged on day 84 with 1 000 pfu of Marburg Musoke virus, IM. All vaccinated animals survived challenge while the three control animals succumbed to infection.
-
The precise correlates of immunity responsible for VLP-elicited protection are not known, but both antibody and T cell responses appear to be important for protection (Warfield K L, et al., 2005). Adoptive transfer of sera or splenocytes from vaccinated animals did not confer protection on naïve mice, but transfer of sera and splenocytes together did provide protection (Warfield K L, et al., 2005). Furthermore, B cell-deficient mice were not protected from challenge after vaccination, suggesting an important role for the antibody response, but T cell deficient mice were also not protected despite the presence of antibody in the vaccinated, T cell-deficient animals (Warfield K L, et al., 2005).
VLP elicit high anti-GP antibody titers in the mouse, guinea pig, and NHP models of infection. At the same time, however, the presence of EBOV-specific antibody titers is not predictive of protection in the small animal models of infection. While both VLP and inactivated EBOV elicited high EBOV-specific IgG titers and high neutralizing antibody titers in mice, only the VLP-vaccinated animals were protected from viral challenge (Warfield K L, et al., 2003). Similarly, animals vaccinated with dose levels that are within ten fold of the protective dose produce comparable anti-GP antibody titers. This has been observed in C57BL/6 mice, Balb/c mice, and Hartley guinea pigs (unpublished data). Critical future studies will look at the relationship between survival and both overall antibody titers and neutralizing antibody titers in NHP; these studies will likely be critical for filovirus vaccine approval under the Food and Drug Administration's (FDA) Animal Rule.
Anti-GP IgG antibody titers typically plateau after the second vaccination. Anti-GP IgG1 and IgG2 isotypes have been observed in mouse, while IgG3 antibody does not seem to be elicited in the absence of adjuvant. IgG3 was observed however, when mice were vaccinated with VLP and the adjuvant QS21 (Warfield K L, et al., 2005). Avidity of the anti-GP antibody is quite high in mice and guinea pigs; sodium thiocyanate treatment of ELISA samples resulted in little decrease in the detected antibody titer, regardless of vaccine dose. Thus there appears to be no correlation between antibody isotype or avidity with either animal survival or vaccine dose using the current assay (unpublished data).
T cell responses to VLP have been observed in C57BL/6 mice, Balb/c mice, and NHP, with both GP and VP40 epitopes eliciting responses from vaccinated animals. The use of adjuvants such as PolyICLC and QS21 greatly enhances the detection of T cell responses. When animals were vaccinated in combination with the adjuvant QS-21, a Th1 profile was observed in cells from vaccinated animals that were re-stimulated with VLP ex vivo (Warfield K L, et al., 2005). IFN-γ secretion was greatly increased by re-stimulation of cells with VLP, but IL4 was not detected, and intracellular staining of cells re-stimulated with overlapping viral peptide pools identified immunodominant epitopes in Balb/c and C57BL/6 mice (Warfield K L, et al., 2005). Data from the vaccination of various knock-out mice suggest that CD8 T cell responses are required for VLP-mediated protection: VLP-vaccinated CD8 T celldeficient mice were not protected from challenge with ma-EBOV while 50% of CD4 T cell-deficient mice were protected (Warfield K L, et al., 2005). Specifically, IFN-γ may be of importance as IFN-γ -deficient animals were also not protected from challenge after vaccination while perforin-deficient animals were protected (Warfield K L, et al., 2005). Interestingly, IgG2b antibody levels were considerably lower in vaccinated CD8-deficient, CD4-deficient, and IFN-γ -deficient animals in comparison to wild type animals, suggesting an avenue for future study.
-
In vitro, VLP are immunogenic and elicit a Th1 cytokine profile from antigen-presenting cells. VLP are taken up by and activate dendritic cells (DC), resulting in an increase in the expression of CD40, HLA-DR, CD80, CD83, and CD86 (Bosio C M, et al., 2004). This is in contrast to live virus, which can infect DCs but which suppresses activation of DCs via the immunosuppressive proteins VP35 and VP24 (Basler C F, et al., 2009; Bosio C M, et al., 2003; Cardenas W B, et al., 2006; Chang T H, et al., 2009; Leung L W, et al., 2011). In addition, VLP treatment results in up-regulation of the chemokine receptor CCR7 on DC. This receptor plays a critical role in DC maturation and migration (Bosio C M, et al., 2004).
Human DCs treated with VLP secreted IL6, IL8, MIP1a, and TNFα; this response was not seen when DCs were treated with inactivated EBOV, nor when VLP were boiled, suggesting that protein structure is critical for eliciting this response (Bosio C M, et al., 2004). IL10 secretion was also observed in bone marrow-derived DCs after VLP treatment (Warfield K L, et al., 2003). Cytokine and NFκB pathway analysis of DCs treated with VLP suggests that the mucin-like domain of GP is particularly important in the immune activation resulting from treatment (Martinez O, et al., 2007).
Similar to the phenotype in DCs, human macrophages up-regulate IL1b, TNFα, IL6, and IL8 mRNA expression within hours of VLP treatment, and TNFα, IL6, and IL8 were detectable in the supernatant within six hours of treatment (Wahl-Jensen V, et al., 2005). Interestingly, this cytokine production was largely driven by the presence of GP, but the results were not replicable with any of the four soluble GP variants tested (Wahl-Jensen V, et al., 2005). Further analysis of VLP-treated human macrophages showed a similar transcriptional profile between treatment with VLP (expressing VP40 and GP) and Ebola virus. This profile was more similar to that of LPS treated cells than to that of cells treated with VLP that only expressed VP40, suggesting again that GP is a critical component of the VLP (Wahl-Jensen V, et al., 2011).
The impact of VLP on NK cells was explored extensively in mice, and it was shown that VLP activate NK cells, resulting in the production of IFN-γ, TNFα, perforin, and other cytokines (Warfield K L, et al., 2004). Intriguingly, VP40 appears to be sufficient for VLP-mediated NK cell activation whereas GP appears to be the critical component for long term immune-mediated protection (Warfield K L, et al., 2004). Early NK responses may be important for initiating the immune response to VLP vaccination.
In vivo studies have explored the immunogenicity of VLP as well. Three days post-vaccination, cells of the mesenteric lymph nodes showed elevated expression of CD25 and CD43 on T cells and CD69 on T and B cells in Balb/c mice (Warfield K L, et al., 2003). VLP vaccination results in no detectable changes in cytokine levels in the peripheral blood of vaccinated mice, suggesting that this vaccine is not eliciting systemic immune activation. However, changes in cytokine and chemokine levels in the lymph node can be detected twenty-four hours after vaccination.
-
The filovirus VLP are a promising vaccine candidate for Ebola and Marburg infection and have the potential to serve as an effective post-exposure therapy as well. In addition, use of VLP as research tools has greatly expanded our understanding of filoviruses. Research using VLP has informed on the nature of the soluble GP products and has provided researchers with a means to study filoviruses in biosaftey level-2 (BSL2) laboratories (Wahl-Jensen V M, et al., 2005). Our understanding of filovirus budding, in particular, has been aided by the development of VLP (Makino A, et al., 2011; Okumura A, et al., 2008; Yasuda J, et al., 2003). The addition of other viral proteins to VLP may expand their utility as a research tool (Johnson R F, et al., 2006).
The primary interest in filovirus VLP, however, lies in their potential as a vaccine. Due to the nature of Ebola and Marburg virus infection and the infrequency of natural outbreaks of infection, vaccine approval will necessarily be by the FDA's animal rule (Sullivan N J, et al., 2009). Among other things, this pathway to vaccine approval requires the identification of immune correlates of protection in an acceptable animal model of infection, which can then translate to humans vaccinated with the candidate vaccine. The difficulty of conducting studies at BSL-4 under the Good Laboratory Practices guidelines is a hurdle that must be overcome. A better understanding of filoviral protection in the NHP model and an understanding of how VLP protect against infection are critical to advancing the development of the VLP vaccine.

 DownLoad:
DownLoad: