









-
Herpes simplex virus-1 (HSV-1) is a double-stranded DNA virus that causes various high-mortality central nervous system (CNS) diseases, including herpes simplex virus encephalitis (HSE) and meningitis (Baringer J R, 2008; Kennedy P G, et al., 2002). HSV-1 has a complex gene structure, and its genetic replication is regulated by host cellular genes. HSV-1 can latently infect the CNS, thereby escaping attacks from the host immune system. It is important to understand the molecular biological processes involved in CNS infection in order to further understand the replication process and pathological mechanisms of HSV-1.
As one of the major cells involved in the immune functions of the CNS, astrocytes play an important role in the pathological processes of HSV-1-induced encephalitis. Astrocytes are involved in host antiviral responses via pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), and HSV-1 infection also affects pathological processes. However, the exact role of astrocytes in pathological infection remains unclear; in particular, little is known about molecular HSV-1 replication in astrocytes.
Our previous studies used siRNA library screening to preliminarily confirm that phosphatase affects HSV-1 replication in astrocytes and bifunctional polynucleotide phosphatase/kinase (PNKP) is a key gene that affects HSV-1 replication. PNKP is a dual-specificity nucleotide phosphatase(Zolner A E, et al., 2011)involved in base excision repair, single-and double-strand break repair, and nonhomologous end joining. (Weinfeld M, et al., 2011). However, the function of PNKP in HSV-1 replication remains unknown. To confirm the mechanisms of PNKP, this study investigates the effects of PNKP on HSV-1 replication. This study also confirms that PNKP inhibits the formation of the circular double-stranded HSV-1 genome, thus affecting viral replication in host cells.
HTML
-
Monkeys (Macaca mulatta, male) were kept in single cages and bred according to the guidelines of the experimental animals committee of the Institute of Medical Biology, Chinese Academy of Medical Sciences (935# Jiaoling, kunming, China)(Gorska P, 2000). We used a single monkey, with a weight of 3 kg and an age of 3.5 years, for each primary culture of astrocytes; astrocyte collection was performed three times in this study. All monkeys were euthanized with sodium pentobarbital. Cerebral samples were obtained 15 minutes after sacrifice.
-
Astrocytes were prepared from the cortices of adult monkeys according to previously described methods(Levison S W, et al., 1991; Norton W T, et al., 1988; Selmaj K W, et al., 1990). Cortices were washed several times with 0.01 mol/L Phosphate Buffered Saline (PBS), cut into small cubes ( < 1 mm3), and digested with 0.25% trypsin for 25 minutes at 37℃. Cell suspensions were sieved through a 40-mm cell strainer. The filtrate was seeded to a density of 1 × 106 cells/cm2 in 100-mm dishes (Corning, NY, USA) and allowed to adhere for 30 minutes in order to easily remove fibroblasts. The plated cells were cultured in a 5% CO2 incubator for 21 days at 37℃, and the culture medium was changed every 5 days as previously described (Frangakis M V, et al., 1984; Tabernero A, et al., 1996). After reaching confluence, the primary cell culture was washed 3× at room temperature with 0.01 mol/L PBS containing 0.25% trypsin. After centrifugation, cells were subsequently cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal calf serum, 100 U/mL penicillin, and 20 μg/mL streptomycin in a 5% CO2 incubator at 37℃. Other glial cells were removed during this procedure, such as oligodendroglia and microglia. The purity of the astrocytes was assessed by the percentage of cells demonstrating glial fibrillary acidic protein (GFAP) fluorescence.
-
RNA oligonucleotides (5'-GGACUCAAGUGGAAC UGGUdTdT-3') were directed against the PNKP sequence. Both the knockdown and negative control siRNAs were designed and synthesized by Guangzhou RiboBio Co., Ltd, Guangzhou, China.
One day before transfection, cells were plated in 2000 µL growth medium without antibiotics in 6-well plates. The monolayers in the 6-well plates were transfected with 50 nmol/L siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
-
Vero cells were grown in Modified Eagle Medium (MEM; GIBCO, Grand Island, NY, USA) supplemented with 10% newborn bovine serum (GIBCO). HSV-1 stocks (8F strain) (Yu X, et al., 2010).
-
The HSV-1 titers in the Vero cells were determined using plaque assays. Cell monolayers were infected with HSV-1 and incubated in 6-well plates containing 700 μL essential medium and 2% Fetal Bovine Serum (FBS). Following virus adsorption, the monolayers were incubated in 2.5 mL essential medium containing 2% FBS and 0.9% agarose. After fixation with 4% formaldehyde (Solarbio, Beijing, China) and the removal of the agarose overlay at 4–5 days postinfection, plaques were visualized by staining with 1% crystal violet (Solarbio) in distilled water.
To analyze the viral growth curves, cells were treated with siRNA for 48 hours. Monolayers were then infected with HSV-1 at 0.01 multiplicity of infection (MOI). Culture supernatants were harvested at 0, 12, 24, 36, 48, 60, 72, 84, and 96 hours postinfection. The viral titers of all samples were determined using the plaque assay.
-
At 72-hours post-siRNA transfection, the astrocyte monolayers were harvested with lysis solution containing 50 mmol/L Tris-HCl (pH 6.8), 0.6% sodium dodecyl sulfate (SDS), 10% glycerol, 1 mmol/L Ethylene Diamine Tetraacetic Acid (EDTA), 1% β-mercaptoethanol, and 0.1% bromphenol blue. The precipitated proteins were quantitated using the BCA method (BCATM, Pierce, Rockford, IL, USA), and equal amounts of protein supernatants were separated on 12% sodium dodecyl sulfate polyacrylamide gel electropheresis (SDS-PAGE) gels. The separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes and detected using antibodies against PNKP (1:500 dilution; SAB4200134; Sigma, St. Louis, MO, USA) and β-actin (1:1000 dilution; Beyotime, Jiangsu, China). After washing 4× with TTBS (0.1% Tween, 0.1 mol/L Tris, pH 8.0, 0.15 mol/L NaCl), the proteins on the PVDF membranes were incubated with HRP-conjugated sheep anti-mouse IgG (1:5000; Huaan, Zhejiang, China). Staining was performed using the ECL Western blot detection reagent (Millipore, Bedford, MA, USA), and β-actin was used as the loading control.
-
Cell viability was determination using the WST-1 cell proliferation assay (Beyotime). The WST-1 reagent was added to the cells at 0, 4, 8, 12, 24, 36, 48, and 72 hours post-transfection, then the cells were incubated for another 2 hours at 37℃. Absorbance was measured at 450 nm using the Synergy-4 Hybrid Microplate Reader (Bio-Tek Instruments, Inc., Winooski, Vt, USA).
-
The astrocyte monolayers were treated with siRNA for 48 hours. Cells were infected with HSV-1 at 8 MOI, then harvested with TRNzol-A+ (Tiangen, Beijing, China) at 2, 4, 6, 8, 12, 16, or 20 hours postinfection. Total RNA was extracted from the infected astrocytes and reverse-transcribed to cDNA using the RevertAidTM First Strand cDNA Synthesis kit (Fermentas, Ontario, Canada).
Real-time polymerase chain reaction (PCR) was used to assess infected-cell proteins 22 (ICP22), thymidine kinase (TK), and Glycoprotein B (gB) mRNA. The results at each time point were used to perform 2-ΔΔCt relative quantitation analysis (β-actin was used as the reference gene).
-
The astrocyte monolayers were treated with siRNA for 48 hours. Cells were infected with HSV-1 (MOI=8). Viruses were harvested with lysis buffer containing 10 mmol/L Tris-HCl, 5 mmol/L EDTA (pH7.5), 0.1% SDS, and 0.2 mg/mL proteinase K at 1, 2, 3, 4, 5, 6, 8, 10, 12, and 14 hours postinfection. Every sample was incubated for 2–3 hours at 37℃. Total DNA was isolated by phenol extraction and ethanol precipitation.
The copy numbers of the ICP0, TK, and GC genes were used to represent the viral copy numbers. The results at each time point were used to perform 2-ΔΔCt relative quantitation analysis (β-actin was used as the reference gene).
-
Astrocyte monolayers were treated with siRNA for 48 hours. Cells were then infected with HSV-1 (MOI = 8). Viruses were harvested at 1, 2, and 5 hours postinfection, and the total DNA of each sample was isolated as described above.
The copy number of the circular fragment of the experimental group relative to control and the copy number of the TK gene were calculated using 2-ΔΔCt relative quantitation analysis (β-actin was used as the reference gene). The ratio of the copy number of the circular fragment to the TK gene represents the ratio of the copy number of the circular viral genome to the total copy number of the genome.
-
Real-time PCR was used to assess ICP0, ICP22, TK, gB, and gC mRNA or DNA using the 7500 Fast Real-time RT-PCR system (Applied Biosystems, Foster City, USA) and the SYBR Green RealMaster kit (Tiangen, Beijing, China). The following primers were used: 5'-GTGCAT GAAAACCTGGATGC-3' and 5'-TTGCCCGTCCAG ATAAAGTC-3' for ICP0; 5'-TGTGCAAGCTTCCTT GTTTG-3' and 5'-GGCATCGGAGATTTCATCAT-3' for ICP22; 5'-CAGCAAGAAGCCACGGAAGT-3' and 5'-GCG TCGGTCACGGCATAA-3' for TK; 5'-CTGGTCAGCTT TCGGTACGA and 5'-CAGGTCGTGCAGCTGTTTGC-3' for gB; 5'-GGTCCACCCTGCCCATTT-3' and 5'-CGG ACGACGTACACGATTGC-3' for GC. β-actin (normalization control) was amplified using 5'-GGCATCCTCACCCTG AAGTA-3' and 5'-GGGGTGTTGAAGGTCTCAAA-3'. The 20-µL total reaction volume contained 9 µL realMasterMix, 200 nmol/L of each primer, and 0.5 µmicro; L DNA. The following protocol was used for these PCR assays: 3 minutes at 95℃ followed by 40 cycles of 95℃ for 10 seconds, 56℃ for 10 seconds, and 68℃ for 40 seconds.
Real-time Taqman PCR assays were performed using the RealMaster Mix kit (Tiangen, Beijing, China) and the 7500 Fast Real-time PCR system (Applied Biosystems) according to the manufacturer's instructions. The 20-µL total reaction volume contained 8 µL RealMasterMix, 200 nmol/L of each primer and the dual-fluorescence dye (FAM/TAMRA)-labeled probe, 1 µL Probe Enhancer solution, and 0.5 µL DNA. The sequences of the circularization-specific primers were the following: forward primer, 5'-GTTGTCGCTGTGAGTTGTGTTGGT-3'; probe, 5'-CGTGCTGTTGGTGTTCTGTTGGTGTT-3'; and reverse primer, 5'-TAGTGCTTGCCTGTCTAACTCGCT-3'. The following protocol was used for these PCR assays: 10 minutes at 95℃, followed by 45 cycles of 95℃ for 10 seconds, 56℃ for 10 seconds, and 68℃ for 90 seconds. β-actin was amplified using the following primers: forward primer, 5'-ACTGAGCGCGGCTACAG-3'; probe, 5'-TTCACCACCACGGCCGAGC-3'; and reverse primer, 5'-CTTAATGTCACGCACGATTTCC-3'
-
Statistical analyses were performed using SPSS 13.0 (SPSS Inc, Chicago, IL, USA). Data obtained from all experiments were evaluated using variance analysis, and p < 0.05 was considered statistically significant.
Animals
Preparation of cerebrocortical astrocytes
siRNA transfection
Cell lines and viruses
Virus assays
Western blot analysis
Cell proliferation assay
Viral gene transcription assay
DNA replication assay
Analysis of the formation of endless genomes
Real-time PCR
Statistical analysis
-
HSV-1 replication in the CNS is a major event in HSE pathology; however, the mechanisms responsible for proliferative infection in neuronal cells remain unclear. Although our previous study found that PNKP might affect the proliferation of HSV-1 in astrocytes, its mechanism also remains unknown. In this study, we used Western blot analysis to examine the role of siRNA in the downregulation of PNKP (Fig. 1). Further analysis of cell proliferation confirmed that siRNA has no significant effect on the proliferation of astrocytes (Fig. 2). These results not only confirm the efficiency and safety of siRNA but also provide fundamental information for future studies on the effects of PNKP on HSV-1 proliferation.
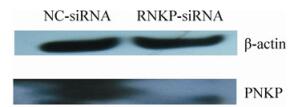
Figure 1. RNAi-mediated knockdown of PNKP. When the cultured primary astrocytes formed a monolayer, the cells were transfected with the nonfunctional siRNA (NC-siRNA) or PNKP-specific siRNA (PNKP-siRNA) (50 nM). Seventy-two hours after transfection, cells were harvested and lysed to collect the total protein. Western blot analysis was performed to detect changes in the PNKP protein levels. β-actin was used as the experimental control.
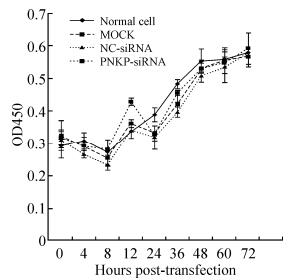
Figure 2. Proliferation of astrocytes was not influenced by siRNA transfection. The negative control siRNA (NC-siRNA) and the PNKP-specific siRNA (PNKP-siRNA) were transfected into the monolayer of primary cultured Rhesus astrocytes. Cells were collected at different time points (0, 4, 8, 12, 24, 36, 48, 60, or 72 hours), and the WST-1 reagent was used to detect cell proliferation. Normal and non-siRNA-transfected cells were used as the experimental controls.
-
To further confirm the effects of PNKP on HSV-1 proliferation and observe the infection characteristics of HSV-1 in PNKP-downregulated astrocytes, this study used siRNA to downregulate PNKP. This was followed by HSV-1 infection (MOI = 0.01) in order to observe any changes in the viral growth curves. PNKP downregulation significantly inhibited viral proliferation (Fig. 3).
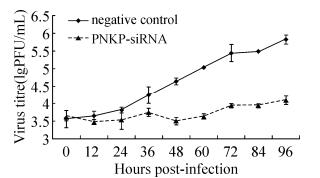
Figure 3. Changes in viral proliferation curves following PNKP downregulation. siRNA (50 nM) was transfected after the primary astrocytes formed a monolayer. Forty-eight hours after transfection, cells were infected with HSV-1 (MOI = 0.01), and the viral solution was collected at different time points (0, 12, 24, 36, 48, 60, 72, 84, or 96 hours). The plaque assay was used to determine the viral titers of each sample, and growth curves were plotted. Cells transfected with nonfunctional siRNA (NC-siRNA) were used as the experimental control.
-
The synthesis of viral proteins was inhibited when HSV-1 proliferation was affected. To determine the influence of viral DNA replication, viral gene transcription—ICP22 (immediate early), TK (early), and gB (late)—was investigated during different stages. PNKP downregulation significantly inhibited the genetic transcription of these viral genes at different stages (Fig. 4).
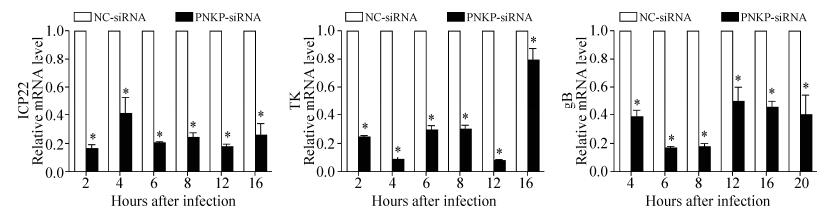
Figure 4. Effect of PNKP downregulation on viral gene transcription following HSV-1 infection. siRNA (50 nmol/L) was transfected after the primary astrocytes formed a monolayer. Forty-eight hours after transfection, cells were infected with HSV-1 (MOI = 8) and the viral solution was collected at different time points (2, 4, 6, 8, 12, 16, or 20 hours). Changes in viral gene transcription during different stages are shown. The results at each time point were used to perform 2-ΔΔCt relative quantitation analysis (β-actin was used as the reference gene; n = 4). The negative control group consisted of cells that were transfected with nonfunctional siRNA (NC-siRNA), and the experimental groups consisted of cells transfected with PNKP-siRNA. *p < 0.05.
-
PNKP downregulation reduced HSV-1 genetic transcription in different stages. We believe that this inhibition occurs after the virus enters the cell and is the initial stage of HSV-1 genome replication (Lehman I R, et al., 1999). In accordance with viral proliferation curve analyses (Fig. 3), we hypothesize that PNKP affects viral genome replication. Here, we used quantitative polymerase chain reaction (qPCR) to detect any changes in the copy number of the viral genome during approximately 1 replication cycle.
Astrocytes were transfected with specific siRNAs that downregulated PNKP, then infected with HSV-1 (MOI = 1) 48 hours later. Within 1 replication cycle, changes in the HSV-1 copy number were detected using qPCR and ICP0, TK, and GC were assessed. When PNKP was not inhibited, the viral copy number peaked 8 hours after infection in comparison with the control group (Fig. 5). When PNKP was downregulated, the viral copy number did not increase and was always at a relatively low level. These results indicate that PNKP downregulation affects the replication of the viral genome in host cells; thus, the viral copy number remained relatively low.
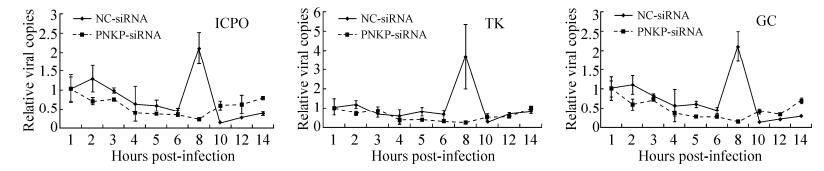
Figure 5. Effect of PNKP downregulation on viral genome copy number after HSV-1 infection. siRNA (50 nM) was transfected after the primary astrocytes formed a monolayer. Forty-eight hours after transfection, the cells were infected with HSV-1 (MOI = 8), and the viral solution was collected at different time points (1, 2, 3, 4, 5, 6, 8, 10, 12, or 14 hours). Changes in the viral copy number at different time points are shown (the copy numbers of the ICP0, TK, and GC genes are used to represent the viral copy numbers). The results at 1 hour and each time point were used to perform 2-ΔΔCt relative quantitation analysis (β-actin was used as the reference gene; n = 4). The negative control group consisted of cells that were transfected with nonfunctional siRNA (NC-siRNA), and the experimental groups consisted of cells that were transfected with PNKP-siRNA.
-
Several models describe HSV-1 replication. The classic model is the θ model: following infection, viral genome cyclization immediately starts (the double-stranded linear genome links head-to-end to form the circular double-stranded genome). θ replication then occurs, and the concatemer structure of the progeny DNA is synthesized by rolling circle replication (Muylaert I, et al., 2011). Using the results of string analysis (http://string-db.org/), we predicted five functional partners (Table 1): LOC712291 (XRCC4), XRCC1, LIG4, C9orf114 and LOC716387. XRCC4, XRCC1 and LIG4 may have relationship with DNA repair according to the descriptions. Interestingly, PNKP, XRCC4, and LigaseIV repair the ends of double-stranded DNA (Weinfeld M, et al., 2011). XRCC4 and LigaseIV are associated with the circular replication of the HSV-1 genome (Muylaert I, et al., 2007). Based on these results, we hypothesize that PNKP plays an important role in the circular replication of the HSV-1 genome. We designed primers that target the two ends of the HSV-1 genome and specifically amplified the junction fragment after genome cyclization as a representation of the copy number of the circular viral genome. We also used the copy number of the TK gene to represent the copy number of the total viral genome. We transfected siRNA into astrocytes to downregulate PNKP and used RT-PCR to detect differences in the ratios of the circular genomes during the early stage of HSV-1 infection.
Name Description Score LOC712291 Similar to X-ray repair cross complementing protein 4 isoform 1 0.894 XRCC1 X-ray repair complementing defective repair in Chinese hamster cells 1 0.815 LIG4 Ligase IV, DNA, ATP-dependent 0.660 C9orf114 Uncharacterized protein C9orf114 0.591 LOC716387 Synovial sarcoma translocation gene on chromosome 18-like 2 0.591 Table 1. Predicted functional partners
Compared with the copy number of the control group, the circular HSV-1 genome demonstrated a significant decrease 1 hour after HSV-1 infection (Fig. 6). This result confirms that PNKP plays an important role in host genome cyclization following HSV-1 infection. PNKP downregulation could affect viral genome cyclization and replication, possibly indicating genome cyclization during virus replication.
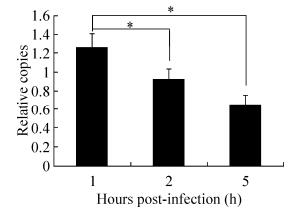
Figure 6. Effect of PNKP on HSV-1 genome cyclization. The copy number of the circular fragment of the experimental group relative to the control group and the copy number of the TK gene were calculated using 2-ΔΔCt relative quantitation analysis (β-actin was used as the reference genes; n = 3). The histogram at each time point shows the ratio of the copy number of the circular fragment to the copy number of the TK gene, which represents the ratio of the copy number of the circular virus genome to the total copy number of the genome. *p < 0.05.
PNKP downregulation by siRNA
Effect of PNKP on HSV-1 proliferation curves
Effect of PNKP downregulation on viral gene transcription following HSV-1 infection
Effect of PNKP downregulation on viral genome copy number after HSV-1 infection
Effect of PNKP on HSV-1 genome cyclization
-
Proliferative HSV-1 infection can be represented using the classic model of genome replication and several prominent features such as genome cyclization, decatenation, and rolling circle replication(Livingston C M, et al., 2008; Muylaert I, et al., 2011). This study used RNA interference to downregulate PNKP and demonstrate that PNKP plays an important role in the cyclization of the HSV-1 double-stranded genome. Our results also confirm that viral genome cyclization occurs within a short period after infection(Garber D A, et al., 1993; Strang B L, et al., 2005), thereby supporting the classic model of HSV-1 replication. In addition to the aforementioned 5'-DNA kinase activity and 3'-DNA phosphatase activity, PKNP also demonstrates adenosine triphosphate (ATP) binding (Jilani A, et al., 1999), nucleic acid binding, protein binding, and transferase activities. To better understand the effects of PNKP on HSV-1, these functions need to be further confirmed using in-depth analyses.
HSV-1 replication is closely associated with numerous macromolecules in host cells and various biological processes, including cellular signal transduction, apoptosis, metabolic activities, and immune responses. Astrocytes play an important role in all of these processes. TLRs and other PRRs play important roles in host antiviral, innate, and adaptive processes that occur in the CNS following HSV-1 infection, including astrocyte(Guo Y, et al., 2011; Pasieka T J, et al., 2011; Reinert L S, et al., 2012). Furthermore, numerous signaling pathways are also activated, including those involving nuclear factor-кB (NF-кb), mitogen-activated protein kinase (MAPK), and interferons. These pathways can initiate the expression of several important inflammation-related factors and chemokines and promote immune responses in astrocytes and other immune cells in the CNS(Li J, et al., 2011; Wuest T R, et al., 2008).
Here, we performed a preliminary analysis of the mechanisms that underlie the effects of PNKP on HSV-1 in astrocytes. We provide data for future studies on the role of astrocytes in the CNS following HSV-1 infection, including immune responses in the CNS and their responses following pathological damage.
-
We are grateful to Huaisheng Shi, Chen Guo, and Jingxian Zhou for capturing the images used in this study. This work was supported by the National Basic Research Program (2012CB518901) (China), National Natural Sciences Foundation of China (31100127) (China), and the Yunnan Natural Science Foundation (2013FZ135, 2013FZ128, 2013FZ134) (China).
-
Conceived and designed the experiments: QH Li; Carried out the experiments: L Yue and SJ Guo; Analyzed the data: L Yue, X Cao and Y Zhang. Contributed reagents and materials: LD Liu and M Yan; Wrote the paper: L Yue, L Sun and QH Li.

 DownLoad:
DownLoad: