









HTML
-
Porcine reproductive and respiratory syndrome (PRRS), caused by the porcine reproductive and respiratory syndrome virus (PRRSV), has been acknowledged as one of the most economically devastating diseases of the swine industry. The genome of PRRSV consists of approximately 15 kilobase pairs (kb)and encodes nine overlapping open reading frames (ORFs), including non-structural protein (Nsp)coding sequence, and structural protein coding sequences ORF2 to ORF7 (Li B et al, 2010; Zhou Y et al, 2012). The non-structural protein, designated as polyprotein, can be divided into Nsp1α, Nsp1β, and Nsp2–Nsp12 (Li B, et al, 2010; Music N, et al, 2010). ORFs 2-7, located upstream of the 3′-UTR (three prime untranslated region), encode viral structural proteins, such as glycoprotein 5 (GP5), glycoprotein 7 (GP7), and nucleocapsid protein (NP) (Li B, et al, 2010; Music N, et al, 2010).
It was reported that a so-called porcine "high fever" disease caused by highly pathogenic PRRSV (HP-PRRSV) emerged in the south of China in 2006, spreading to more than 10 provinces (autonomous cities or regions) and affecting over two million pigs, with about 400, 000 fatal cases (Tian K, et al, 2007). However, research was lacking as to whether the HP-PRRSV isolates were dominant during 2007-2012. Considering the extensive use of PRRSV vaccines for preventing and controlling PRRS in mainland China, the appearance of a natural recombination event between a vaccine strain and a field isolate is noteworthy. In the present study, 89 PRRSV isolates in China during 2007 to 2012 were selected from the GenBank genetic sequence database. The evolutionary characteristics of these isolates were analyzed based on the sequences of Nsp2 and GP5. The genetic variations of the isolates were also compared with six representative strains. The results of the present study may prove useful for the epidemiology of PRRSV as well as for vaccine development.
-
In China, between 2007 and 2012, 89 PRRSV isolates were collected from the GenBank database (Supplementary Table 1). Six strains were selected as representative strains, including the representative American-type PRRSV vaccine strain VR2332, three vaccines strains used in China (CH-1R, MLV RespPRRS/Repro, and SP), and two HP-PRRSV isolates (GD and JXA1). The ClustalW (multiple sequence alignment) program (Thompson J D, et al, 1994) was used to generate an alignment file for these PRRSV isolates.
-
Phylogenetic and molecular evolutionary genetic analyses were conducted by using the distance-based neighbor-joining method with MEGA 5.1 software (Kumar S et al, 2004). Bootstrap values were calculated from 1, 000 replicates of the alignment.
-
Recombination analysis was performed by comparing full-length genome sequences of the isolates using a recombination detection program (RDP), as described by Martin D P, et al (2010). Recombinant region and parental sequences were identified by five procedures (BootScan, Chimera, GENECONV, MaxChi, and RDP) integrated in an RDP4 software package, with correction for multiple comparisons (P < 0.001).
Isolates
Phylogenetic analysis
Recombination analysis
-
Nsp2 is the largest non-structural protein cleavage from a polyprotein (Snijder E J, et al, 1995; Zhang M, et al, 2013). Nsp2 has been recognized as the most variable region within the PRRSV genome (Allende R, et al, 1999; Yoshii M, et al, 2008; Zhou Y, et al, 2012; Wang F X, et al, 2013). The hypervariable region of Nsp2 in HP-PRRSV isolates contained a discontinuous deletion equivalent to 30 amino acids (Tian K, et al, 2007). Nsp2 is considered an important region for monitoring the genetic and epidemiological evolution of PRRSV (Wang F X, et al, 2013). In this study, the deduced amino acid (aa) sequences of 89 complete Nsp2 proteins of PRRSV exhibited different lengths, indicating that a great variety of PRRSV isolates exist in China (Supplementary Table 1). The length of amino acids ranged from 1022 aa to 1230 aa. There were 601 (48.9%) variable sites, including singleton sites (13.1%) and parsim-informative sites (35.8%). These data suggested that a high degree of genetic diversity existed among the PRRSV population in China. Out of 89 Nsp2 proteins, 76 (85.4%) had the same length of 1166 aa. Furthermore, 80 out of 89 (88.9%) Nsp2 proteins had a discontinuous deletion of 30 amino acids in Nsp2, indicating that isolates with the discontinuous 30-aa deletion in the Nsp2 region represented the dominant virus during 2007-2012.
To explore the relationship between these isolates and six representative strains, a phylogenetic tree of the Nsp2 was constructed by using the distance-based neighbor-joining method with MEGA4 software. The results showed that the 89 isolates could be divided into three subgroups (Figure 1): subgroup Ⅰ, Ⅱ, and Ⅲ. The majority of these isolates were grouped into subgroup Ⅰ, whose representatives isolates were GD, JXA1, and vaccine strain CH-1R. Subgroup Ⅱ was composed of GM, QYYZ, and QY2010. Finally, subgroup Ⅲ was composed of DY, YN-2011, and three representative vaccine strains (VR2332, SP, and MLV RespPRRS/Repro). The sequence comparison showed that the Nsp2 of DY, YN-2011, and HH08 had 83.61%-99.67%, 82.94%-98.83%, and 78.73%-98.49% homologous identity with that of vaccine strains at an amino acid level (Table 1). Isolates DY and YN-2011 had the greatest homology with MLV RespPRRS/Repro, while isolate HH08 exhibited the greatest homology with CH-1R. Furthermore, comparing the Nsp2 aa sequence of DY with that of MLV RespPRRS/Repro, mutations were observed at position T530→A530, E540→G540, K569→E569, and K636→E636. Isolate HH08 had 98.49% homology to CH-1R, with 18 mutated sites in Nsp2. These results suggested that HP-PRRSV isolates represented the dominant virus during 2007-2012. Three isolates–HH08, DY, and YN-2011–were more closely related to the vaccine strains.
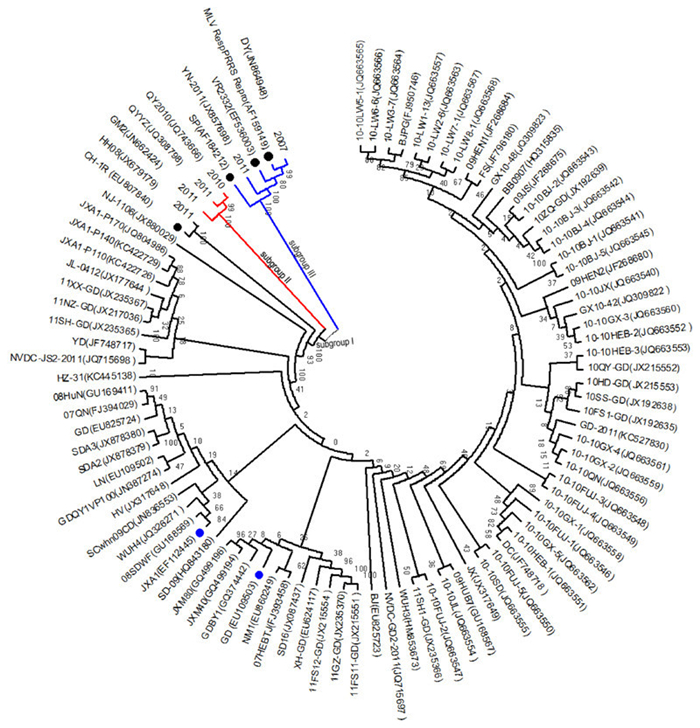
Figure 1. Phylogenetic tree representing 89 isolates during 2007-2012 and six representative strain sequences based on the Nsp2 genes. The phylogenetic tree was constructed by the distance-based neighbor-joining method using MEGA5.1 software. Bootstrap values were calculated from 1, 000 replicates of the alignment. Different subgenotypes were indicated by different colors. Black branches indicate subgroup Ⅰ, red branches indicate subgroup Ⅱ, and blue branches indicate subgroup Ⅲ. Six representative strains, including the representative American-type PRRSV vaccine strain VR2332 (EF536003), three vaccine strains used in China [CH-1R(EU807840), MLV RespPRRS/Repro (AF159149), and SP(AF184212)], and two HP-PRRSV isolates [GD(EU109503) and JXA1 (EF112445)], are indicated by filled circles. Black-filled circles indicate vaccine strains, while blue-filled circles indicate HP-PRRSV isolates.
Species 1 Species 2 Nsp2 identity
(%)ORF5 identity
(%)VR2332 DY 99.16 98.5 MLV RespPRRS/Repro DY 99.67 100 CH-1R DY 83.61 89 SP DY 84.17 92 VR2332 YN-2011 98.49 97.5 MLV RespPRRS/Repro YN-2011 98.83 99 CH-1R YN-2011 82.94 88 SP YN-2011 83.77 91 VR2332 HH08 83.19 89 MLV RespPRRS/Repro HH08 83.44 88.5 CH-1R HH08 98.49 97.5 SP HH08 78.73 92.5 * Underscored numbers indicate the highest identities between species 1 and 2, respectively. Table Table 1. List of flavivirus NS2BNS3 structures
-
Glycoprotein 5 (GP5) encoded by ORF5 is one of the major structural proteins. It comprises 2-4 glycosylation sites, a 31-aa signal peptide, and 6 antigenic determinants that induce neutralizing antibodies (Yin G, et al, 2009; Zhou L, et al, 2009; Mu C, et al, 2013). GP5 is one of the structural proteins with the highest variability in PRRSV (Zhang M, et al, 2013). As the main candidate protein for the development of vaccine subunits, also as a result of its immunological significance and polymorphic nature, GP5 has been the target for analysis of the genetic diversity of PRRSV (Yin G, et al, 2009). The results in this study showed that the length of deduced amino acids of 89 complete ORF5 of PRRSV ranged from 199 aa to 201 aa (Supplementary Table 1). Out of 89 GP5 proteins, 86 had the same length of 200 amino acids (Supplementary Table 1). Isolate GD-2011 had 201 aa, with N60 insertion. Isolates 07QN, 09JS, and JX had 199 aa, with L200 deletion. There were 83 (41.2%) variable sites, including singleton sites (14.3%) and parsim-informative sites (26.9%). The functional domains of GP5–such as the signal peptide, ectodomain, transmembrane regions, endodomain, primary neutralizing epitope (PNE), and decoy epitope–were analyzed by Zhou Y et al (2009). Compared with the representative vaccine strains, the mutation sites of 89 isolates were located at position 37, 38, 39, 41, 43, and 52 of the PNE (Supplementary Table 1). The most variable site of the PNE was L39/F39, including L39/F39→S39 (4 isolates)and L39/F39→I39 (83 isolates). Other representative amino acid mutations of the PNE (S37→P37, H38→Q38/Y38, L41→S41, Y43→C43, and G52→V52) were also found in these 89 isolates. It has been reported that ORF5 codon positions 13 and 151 played a key role in denaturing VR2332 to MLV RespPRRSV (Yin G, et al, 2009). In this study, Q13 was observed in 7 of 89 isolates (7.87%), while R13 was observed in the remaining 82 isolates (92.13%) (Supplementary Table 2). A conserved G151 was observed in 89 isolates (Supplementary Table 2). Interestingly, the GP5 of isolates DY, YN-2011, and HH08 was highly homologous to four representative vaccine strains–VR2332, CH-1R, SP, and MLV RespPRRS/Repro–with 89%-100%, 88%-99%, and 88.5%-97.5% identities, respectively (Table 1). Isolates DY and YN-2011 had the greatest homology with MLV RespPRRS/Repro, while isolate HH08 exhibited the greatest homology with CH-1R. It could then be concluded that a recombination might have occurred between these strains.
-
Studies have shown that recombination events have occurred in PRRSV (van Vugt J, et al, 2001; Yuan S, et al, 1999; Liu D, et al, 2011; Lu W, et al, 2012; Chen N, et al, 2013; Shi M, et al, 2013). Lu W, 2012reported that isolate GM2 was a potential recombinant between the MLV RespPRRS/Repro vaccine strain and the Chinese field strain QYYZ. These events might have major implications for virus evolution, pathogenicity, vaccine safety and efficiency, or diagnosis. To confirm whether recombination occurred between these isolates, recombinant analysis was conducted based on the fulllength genome sequences of PRRSV isolates, using RDP4 software, as described byMartin D, 2010. The results showed that the recombination sites were located in positions 3345-3707 and 12269-12981 (DY), or positions 3345-3707 and 12269-12955 (YN-2011) of the coding region of PRRSV isolates, with a significance level of p < 0.001 (Figure 2). The recombinant fragment 3345-3707 was located in the Nsp2 coding region, while the recombinant fragment 12269-12981 (or 22269-12955) was located in the GP2 and GP3 coding regions. The strain SP was identified as the major parent, while the strain CH-1R was the minor parent(Figure 2). These results indicated that both isolates, namely YN-2011 and DY, were the evolutionary products of recombination events between SP and CH-1R. It has been shown that recombination was an important genetic mechanism, contributing to the variation and evolution of PRRSV, and that HP-PRRSV was a variant of Type 2 PRRSV with high virulence (Liu D, et al, 2011; Chen N, et al, 2013). The results in this study further proved that extensive use of attenuated PRRSV vaccine also contributed to the increasing diversity of PRRSV in the field. In addition, mutations potentially correlated with the overattenuation of an HP-PRRSV strain (Yu X, et al, 2013). The antigenicity and immunogenicity of overattenuated vaccine were significantly reduced during the overattenuation (Yu X, et al, 2013). As recombination requires the co-infection of a pig with more than one PRRSV isolate, restrictions on usage of attenuated vaccine could help to reduce risks induced by recombination. Furthermore, the use of inactivated vaccines, which are currently available, is also problematic since they do not induce effective protective immunity. We strongly recommend that priority should be given to the development and use of other vaccines, such as genetic vaccines and subunit vaccines. Moreover, the focus of attention should be directed toward measures to improve the immunogenicity of inactivated vaccine.

Figure 2. Recombination plots of the coding regions from selected sequences. Recombination analysis was performed by comparing full-length genome sequences of isolates DY, YN-2011, and HH08 with four vaccine strains (VR2332, CH-1R, SP, and MLV RespPRRS/Repro). Recombinant region and parental sequences were identified using five procedures (Bootscan, Chimera, GENECONV, MaxChi, and RDP)integrated in an RDP4 software package with correction for multiple comparisons (P < 0.001). Nucleotide positions of detected breakpoints are indicated
HP-PRRSV isolates were the dominant virus isolates during 2007-2012
Recombination occurred due to extensive use of the representative vaccine strains
Extensive use of attenuated PRRSV vaccine contributed to the increasing diversity of PRRSV in the field
-
In summary, the results in this study have suggested that a high degree of genetic diversity has existed among the PRRSV population in China. HP-PRRSV isolates, with a discontinuous 30-aa deletion in Nsp2 region, were still the dominating virus during 2007-2012. Owing to the extensive use of the representative vaccine strains, recombination events occurred. Three isolates, namely HH08, DY and YN-2011, were more closely related to vaccine strains. Both the YN-2011 and DY isolates were the evolutionary products of recombination events between SP and CH-1R. The development and usage of attenuated vaccines for the disease should be reconsidered because of recombination between different isolates in the field.
-
This work was supported by the Jilin Province Science and Technology Development Project (No. 20140101123 JC), the Fundamental Research Fund of Jilin University, and the Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1248).
-
The authors declare that they have no conflicts of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
RLZ designed the experiments. CYH performed the experiments. OYHS, ZMJ, CFW, and YX analyzed the data. RLZ and PD wrote the paper. All authors read and approved the final manuscript.
Supplementary tables are available on the website of Virologica Sinica: http://www.virosin.org.

 DownLoad:
DownLoad: